| The function of the heart is to pump blood through the circulatory system, and this is accomplished by the highly organized contraction of cardiac muscle cells. Specifically, the cardiac muscle cells are connected together to form an electrical syncytium, with tight electrical and mechanical connections between adjacent cardiac muscle cells. An action potential initiated in a specialized region of the heart (e.g., the sinoatrial node) is therefore able to pass quickly throughout the heart to facilitate synchronized contraction of the cardiac muscle cells, which is important for the pumping action of the heart. Likewise, refilling of the heart requires synchronized relaxation of the heart, with abnormal relaxation often resulting in pathological conditions.
|
| In this chapter attention is initially directed at the organization of cardiac muscle cells within the heart, including discussion of the tight electrical and mechanical connections. The mechanisms underlying contraction, relaxation, and regulation of the force of contraction of cardiac muscle cells are also addressed. It is noteworthy that although cardiac muscle and skeletal muscle are both striated muscles, there are significant differences in terms of organization, electrical and mechanical coupling, excitation-contraction coupling, and mechanisms to regulate the force of contraction. These differences are also highlighted.
|
| BASIC ORGANIZATION OF CARDIAC MUSCLE CELLS
|
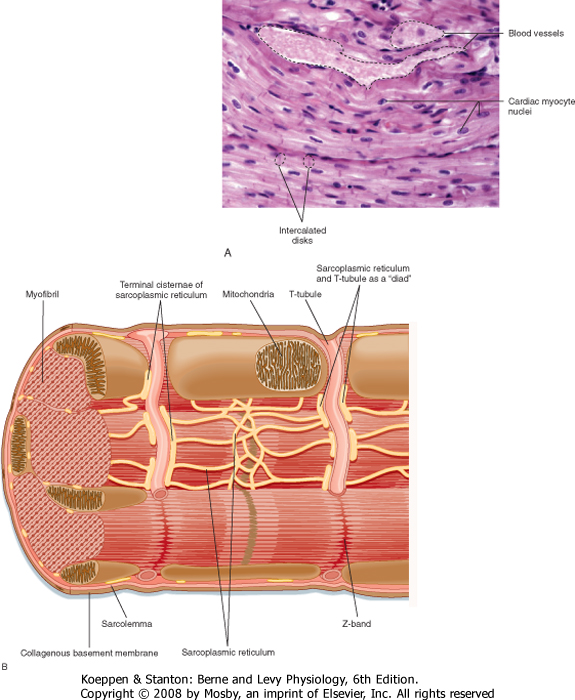
| Cardiac muscle cells are much smaller than skeletal muscle cells. Typically, cardiac muscle cells measure 10 μm in diameter and approximately 100 μm in length. As shown in Figure 13-1, A, cardiac cells are connected to each other through intercalated disks, which include a combination of mechanical junctions and electrical connections. The mechanical connections, which keep the cells from pulling apart when contracting, include the fascia adherens and desmosomes. Gap junctions between cardiac muscle cells, on the other hand, provide electrical connections between cells to allow propagation of the action potential throughout the heart. Thus, the arrangement of cardiac muscle cells within the heart is said to form an electrical and mechanical syncytium that allows a single action potential (generated within the sinoatrial node) to pass throughout the heart so that the heart can contract in a synchronous, wave-like fashion. Blood vessels course through the myocardium.
|
| The basic organization of thick and thin filaments in cardiac muscle cells is comparable to that seen in skeletal muscle (see Chapter 12). When viewed by electron microscopy, there are repeating light and dark bands that represent I bands and A bands, respectively (Fig. 13-1, B). Thus, cardiac muscle is classified as a striated muscle. The Z line transects the I band and represents the point of attachment of the thin filaments. The region between two adjacent Z lines represents the sarcomere, which is the contractile unit of the muscle cell. The thin filaments are composed of actin, tropomyosin, and troponin and extend into the A band. The A band is composed of thick filaments, along with some overlap of thin filaments. The thick filaments are composed of myosin and extend from the center of the sarcomere toward the Z lines.
|
| Myosin filaments are formed by a tail-to-tail association of myosin molecules in the center of the sarcomere, followed by a head-to-tail association as the thick filament extends toward the Z lines. Thus, the myosin filament is polarized and poised for pulling the actin filaments toward the center of the sarcomere. A cross section of the sarcomere near the end of the A band shows that each thick filament is surrounded by six thin filaments and each thin filament receives cross-bridge attachments from three thick filaments. This complex array of thick and thin filaments is characteristic of both cardiac and skeletal muscle and helps stabilize the filaments during muscle contraction (see Fig. 12-3, B, for the hexagonal array of thick and thin filaments in the sarcomere of striated muscle).
|
| There are several proteins that may contribute to the organization of the thick and thin filaments, including meromyosin and C protein (in the center of the sarcomere), which appear to serve as a scaffold for organization of the thick filaments. Similarly, nebulin extends along the length of the actin filament and may serve as a scaffold for the thin filament. α-Actinin anchors the actin filament to the Z line, whereas the protein tropomodulin resides at the end of the actin filament and regulates the length of the thin filament. These proteins are present in both cardiac and skeletal muscle cells.
|
| page 256 |  | | page 257 |
| Figure 13-1 A, Photomicrograph of cardiac muscle cells (210×). Intercalated disks at either end of a muscle cell are identified in the lower left portion of the micrograph. The intercalated disk physically connects adjacent myocytes and, because of the presence of gap junctions, electrically couples the cells as well so that the muscle functions as an electrical and mechanical syncytium. B, Schematic representation of the organization of a sarcomere within a cardiac muscle cell. (A, From Telser A: Elsevier's Integrated Histology. St. Louis, Mosby, 2007; B, redrawn from Fawcett D, McNutt NS: J Cell Biol 42:1-45, 1969.) |
| The thick filaments are tethered to the Z lines by a large elastic protein called titin. Although titin was postulated to tether myosin to the Z lines and thus prevent overstretching of the sarcomere, there is evidence indicating that titin may participate in cell signaling (perhaps by acting as a stretch sensor and thus modulating protein synthesis in response to stress). Such signaling by titin has been observed in both
cardiac and skeletal muscle cells. Moreover, genetic defects in titin result in atrophy of both cardiac and skeletal muscle cells and may contribute to both cardiac dysfunction and skeletal muscle dystrophies (recently termed titinopathies). Titin is also thought to contribute to the ability of cardiac muscle to increase force upon stretch (discussed later).
|
| page 257 | | | page 258 |
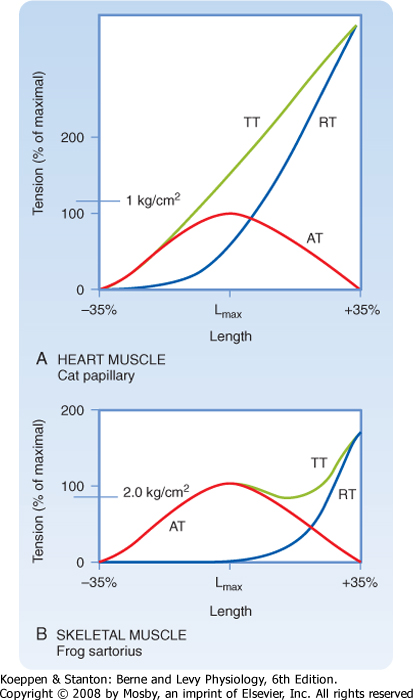
| Figure 13-2 Cardiac muscle (panel A) has high resistance to stretch when compared with skeletal muscle (panel B). When either cardiac or skeletal muscle is stretched, there is an increase in resting tension (RT). If the muscle is then stimulated to contract maximally, it generates more tension (termed total tension-TT). The difference between total tension and resting tension at any given length is the force produced by contraction (e.g., active tension-AT). The bell-shaped dependence of active tension on muscle length is consistent with the sliding filament theory of cardiac and skeletal muscle. It is, however, difficult to stretch cardiac muscle beyond its optimal sarcomere length, as evidenced by the rapid rise in resting tension in the middle of the bell-shaped AT curve. |
| Although cardiac muscle and skeletal muscle both contain an abundance of connective tissue, there is more connective tissue in the heart. The abundance of connective tissue in the heart helps prevent muscle rupture (as in skeletal muscle), but it also prevents overstretching of the heart. Length-tension analysis of cardiac muscle, for example, shows a dramatic
increase in passive tension as cardiac muscle is stretched beyond its resting length (Fig. 13-2). Skeletal muscle, by contrast, tolerates a much greater degree of stretch before passive tension increases to a comparable level. The reason for this difference between cardiac and skeletal muscle is not known, although one possibility is that stretch of skeletal muscle is typically limited by the range of motion of the joint, which in turn is limited by the ligaments/connective tissue surrounding the joint. The heart, on the other hand, appears to rely on the abundance of connective tissue around cardiac muscle cells to prevent overstretching
during periods of increased venous return. During intense exercise, for example, venous return may increase fivefold. However, the heart is capable of pumping this extra volume of blood into the arterial system with only minor changes in the ventricular volume of the heart (i.e., end-diastolic volume increases less than 20%). Although the abundance of connective tissue in the heart limits stretch of the heart during these periods of increased venous return, additional regulatory mechanisms help the heart pump the extra blood that it receives (as discussed later). Conversely, if the heart were to be overstretched, the contractile ability of cardiac muscle cells would be expected to decrease (because of decreased overlap of the thick and thin filaments), thereby resulting in insufficient pumping, increased venous pressure, and perhaps pulmonary edema.
|
| Within cardiac muscle cells, myofibrils are surrounded by the sarcoplasmic reticulum (SR), an internal network of membranes (Fig. 13-1, B). This is similar to skeletal muscle except that the SR in the heart is less dense and not as well developed. Terminal regions of the SR abut the T tubule or lie just below the sarcolemma (or both) and play a key role in the elevation of intracellular [Ca++] during an action potential. The mechanism by which an action potential initiates release of Ca++ in the heart, however, differs significantly from that in skeletal muscle (as discussed later). The heart contains an abundance of mitochondria, with up to 30% of the volume of the heart being occupied by these organelles. The high density of mitochondria provides the heart with great oxidative capacity, more so than typically seen in skeletal muscle.
|
| Familial cardiomyopathic hypertrophy (FCH) occurs in approximately 0.2% of the general population but is a leading cause of sudden death in otherwise healthy adults. It has been linked to genetic defects in a variety of proteins in cardiac sarcomeres, including myosin, troponin, tropomyosin, and myosin-binding protein C, a structural protein located in the middle of the A band of the sarcomere. FHC is an autosomal dominant disease, and transgenic studies indicate that expression of only a small amount of the mutated protein can result in development of the cardiomyopathic phenotype. Moreover, mutation of a single amino acid in the myosin molecule is sufficient to produce cardiomyopathic hypertrophy. The pathogenesis of FHC, however, is variable, even within a family with a single gene defect, both in terms of onset and severity, thus suggesting the presence of modifying loci. |
| page 258 | | | page 259 |
| The sarcolemma of cardiac muscle also contains invaginations (T tubules), comparable to those seen in skeletal muscle. In cardiac muscle, however, T tubules are positioned at the Z lines, whereas in mammalian skeletal muscle, T tubules are positioned at the ends of the I bands. In cardiac muscle there also tends
to be fewer and less well developed connections between the T tubules and the SR than in skeletal muscle.
|
| CONTROL OF CARDIAC MUSCLE ACTIVITY
|
| Cardiac muscle is an involuntary muscle with an intrinsic pacemaker. The pacemaker represents a specialized cell (located in the sinoatrial node of the right atrium) that is able to undergo spontaneous depolarization and generate action potentials. It is important to note that although several cells in the heart are able to depolarize spontaneously, the fastest spontaneous depolarizations occur in cells in the sinoatrial node. Moreover, once a given cell spontaneously depolarizes and fires an action potential, this action potential is then propagated throughout the heart (by specialized conduction pathways and cell-to-cell contact). Thus, depolarization from only one cell is needed to initiate a wave of contraction in the heart (i.e., heartbeat). The mechanism or mechanisms underlying this spontaneous depolarization are discussed in depth in Chapter 16.
|
| As shown in Figure 16-17, once an action potential is initiated in the sinoatrial node, it is propagated between atrial cells via gap junctions, as well as through specialized conduction fibers in the atria. The action potential can pass throughout the atria within approximately 70 msec. For the action potential to reach the ventricles, it must pass through the atrioventricular node, after which the action potential passes throughout the ventricle via specialized conduction pathways (bundle of His and Purkinje system) and gap junctions in the intercalated disks of adjacent cardiac myocytes. The action potential can pass through the entire heart within 220 msec after initiation in the sinoatrial node. Because contraction of a cardiac muscle cell typically lasts 300 msec, this rapid conduction promotes nearly synchronous contraction of heart muscle cells. This is a very different scenario from that of skeletal muscle, where cells are grouped into motor units that are recruited independently as the force of contraction is increased.
|
| Excitation-Contraction Coupling
|
| Blood and extracellular fluids typically contain 1 to 2 mM free Ca++, and it has been known since the days of the physiologist Sidney Ringer (ca. 1882) that the heart requires extracellular Ca++ to contract. Thus, an isolated heart typically continues to beat when perfused with a warm (37° C), oxygenated, physiological salt solution that contains approximately 2 mM Ca++ (e.g., Tyrode's solution), but it stops beating in the absence of extracellular Ca++. This cessation of contractions in Ca++-deficient media is also observed in hearts that are electrically stimulated, thus further demonstrating the importance of extracellular Ca++ for contraction of cardiac muscle. This situation is quite different from that of skeletal muscle, which can contract in the total absence of extracellular Ca++.
|
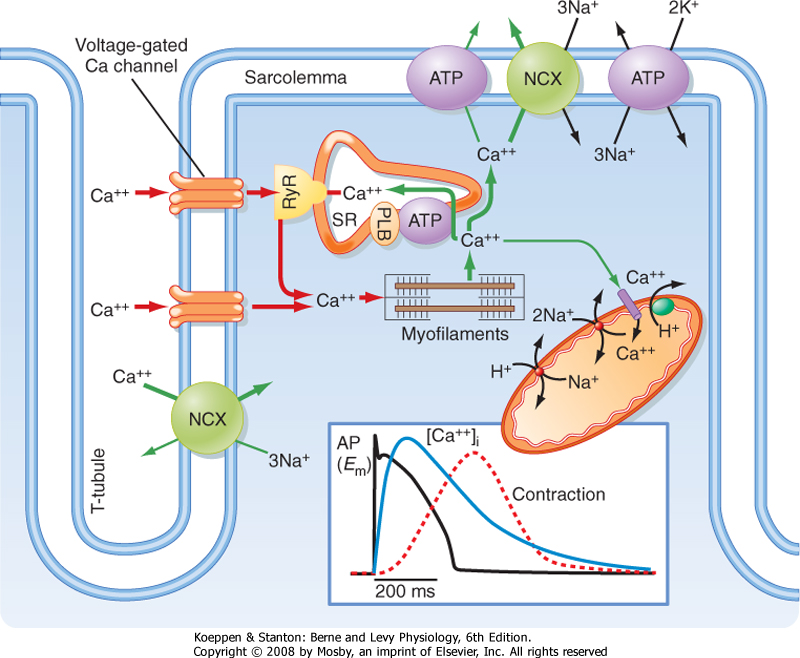
|
| Figure 13-3 Excitation-contraction coupling in the heart requires Ca++ influx through L-type Ca++ channels in the sarcolemma and T tubules. See text for details. (Redrawn from Bers DM: Nature 415:198-205, 2002.) |
| page 259 | | | page 260 |
| Examination of the action potential in cardiac muscle reveals a prolonged action potential lasting 150 to 300 msec (Fig. 13-3), which is substantially longer than the action potentials in skeletal muscle (≈5 msec). The long action potential duration in cardiac muscle is due to a slow inward Ca++ current
through a voltage-gated L-type Ca++ channel in the sarcolemma. The amount of Ca++ coming into the cardiac muscle cell is relatively small and serves as a trigger for release of Ca++ from the SR. In the absence of extracellular Ca++, one is still able to initiate an action potential in cardiac muscle, although it is considerably shorter in duration and unable to initiate a contraction. Thus, influx of Ca++ during the action potential is critical for triggering release of Ca++ from the SR and thus initiating contraction.
|
| The L-type Ca++ channel is composed of five subunits (α1, α2, β, γ, and δ). The α1 subunit is also called the dihydropyridine receptor (DHPR) because it binds the dihydropyridine class of Ca++ channel blocking drugs (e.g., nitrendipine and nimodipine). Although this channel complex is present in both skeletal and cardiac muscle, it serves very different functions in the two muscle types (see later).
|
| In each cardiac muscle sarcomere, terminal regions of the SR abut T tubules and the sarcolemma (Figs. 13-1, B, and 13-3). These junctional regions of the SR are enriched in ryanodine receptors (RYRs; an SR Ca++ release channel). The RYR is a Ca++-gated Ca++ channel, so influx of Ca++ during an action potential is able to initiate release of Ca++ from the SR in cardiac muscle. The amount of Ca++ released into the cytosol from the SR is much greater than that entering the cytosol from the sarcolemma, although release of Ca++ from the SR does not occur without this entry of "trigger" Ca++. This contrasts with skeletal muscle, where release of Ca++ from the SR does not involve entry of Ca++ across the sarcolemma but instead results from a voltage-induced conformational change in the DHPR. Thus, excitation-contraction coupling in cardiac muscle is termed electrochemical coupling (involving Ca++-induced release of Ca++), whereas excitation-contraction coupling in skeletal muscle is termed electromechanical coupling (involving direct interactions between the DHPR in the T tubule and the RYR in the SR). The basis for this difference in Ca++ release mechanisms appears to depend on the DHPR isoform because expression of cardiac DHPR in skeletal muscle cells results in a requirement for extracellular Ca++ for contraction of these modified skeletal muscle cells.
|
| As in skeletal muscle, contraction of cardiac muscle is thin filament regulated, with an elevation in intracellular [Ca++] required to promote actin-myosin interaction. At low (<50 nM) intracellular [Ca++], binding of myosin to actin is blocked by tropomyosin. As cytosolic [Ca++] increases during an action potential, however, binding of Ca++ to troponin C results in a conformational change in the troponin/tropomyosin complex such that tropomyosin slips into the groove of the actin filament and exposes myosin binding sites on the actin filament. As long as cytosolic [Ca++] remains elevated and hence myosin binding sites are exposed, myosin will bind to actin, undergo a ratchet action, and contract the cardiac muscle cell. Note that because myosin binding sites on actin are blocked at low [Ca++] and exposed during a rise in intracellular [Ca++], contraction of cardiac muscle is termed "thin filament regulated." This is identical to the situation in skeletal muscle but contrasts with smooth muscle, where contraction is thick filament regulated (see Chapter 14).
|
| During a rise in intracellular [Ca++] and exposure of myosin binding sites on actin, the myosin cross-bridges undergo a series of steps resulting in contraction of the cardiac muscle cell. At rest, the myosin molecules are energized in that they have partially hydrolyzed ATP to "cock the head" and are thus ready to interact with actin. An elevation in intracellular [Ca++] then exposes myosin binding sites on actin and thus allows myosin to bind actin (step 1). The bound myosin subsequently undergoes a power stroke in which the actin filament is pulled toward the center of the sarcomere (step 2). ADP and Pi are released from the myosin head during this step as the energy from ATP is used to contract the muscle. The myosin head moves approximately 70 nm during each ratchet action (cross-bridge cycle). Binding of ATP to myosin decreases the affinity of myosin for actin and thus allows myosin to release from actin (step 3). Myosin then partially hydrolyzes the bound ATP to reenergize ("cock") the head (step 4) and ready the cross-bridge for another cycle. This four-step cycle is identical to that described for skeletal muscle (see Chapter 12).
|
| Cardiac muscle and skeletal muscle differ, however, in the level of intracellular [Ca++] attained after an action potential and hence in the number of actin-myosin interactions. In skeletal muscle, the rise in intracellular [Ca++] and the number of actin-myosin interactions are high after an action potential. In cardiac muscle, the rise in intracellular [Ca++] can be regulated, which affords the heart an important means of modulating the force of contraction without recruiting more muscle cells or undergoing tetany. Recall that in the heart all the muscle cells are activated during a contraction, so recruiting more muscle cells is not an option. Moreover, tetany of cardiac muscle cells would prevent any pumping action and thus be fatal. Consequently, the heart relies on different means of increasing the force of contraction, including varying the amplitude of the intracellular Ca++ transient.
|
| Relaxation of Cardiac Muscle
|
| page 260 | | | page 261 |
| Relaxation of skeletal muscle simply requires reaccumulation of Ca++ by the SR through the action of the SR Ca++ pump (SERCA). Although SERCA plays a key role in the decrease in cytosolic [Ca++] in cardiac muscle, the process is more complex than that in skeletal muscle because some trigger Ca++ enters the cardiac muscle cell through the sarcolemmal Ca++ channels during each action potential. A mechanism must therefore exist to extrude this trigger Ca++; otherwise, the amount of Ca++ in the SR would continuously increase and result in Ca++ overload. In particular, some Ca++ is extruded from the cardiac muscle cell though the sarcolemmal 3Na+-1Ca++ antiporter and a sarcolemmal Ca++ pump (Fig. 13-3). Note that extracellular [Ca++] is in the millimolar range whereas intracellular [Ca++] is submicromolar, so extrusion of Ca++ is
accomplished against a large chemical gradient. Similarly, [Na+] is considerably higher in the extracellular media than within the cell. The antiporter uses the Na+ gradient across the cell to power the uphill movement of Ca++ out of the cell. Because 3 Na+ ions enter the cell in exchange for 1 Ca++ ion, the 3Na+-1Ca++ antiporter is electrogenic and creates a depolarizing current. The sarcolemmal Ca++ pump, on the other hand, uses the energy in ATP to extrude Ca++ from the cell. Both extrusion mechanisms and SERCA thus contribute to the relaxation of cardiac muscle by decreasing cytosolic [Ca++].
|
| Although the interaction of actin and myosin requires a relatively small increase in free intracellular [Ca++], the abundance of Ca++-binding proteins in the myoplasm necessitates a much larger increase in total intracellular [Ca++]. The resting intracellular [Ca++] is approximately 50 to 100 nM, with half-maximal force of contraction requiring approximately 600 nM free Ca++ (Fig. 13-4). However, because of Ca++-binding proteins such as parvalbumin and troponin C, the total myoplasmic concentration must increase by 70 μM. As already noted, much of this increase in total myoplasmic [Ca++] occurs through release of Ca++ from the SR. In a number of species, including rabbits, dogs, cats, guinea pigs, and humans, uptake and release of Ca++ by the SR account for approximately 70% of the intracellular Ca++ transient. Thus, up to 30% of the rise in intracellular [Ca++] may be attributable to influx of Ca++ through voltage-gated Ca++ channels in the sarcolemma, with the 3Na+-1Ca++ antiporter contributing significantly to Ca++ extrusion during relaxation.
|
| The sarcolemmal Ca++ pump is in lower abundance than the 3Na+-1Ca++ antiporter but has a higher affinity for Ca++ and thus may contribute more to the regulation of resting intracellular [Ca++] (Fig. 13-4). The relative contribution of the Ca++ extrusion mechanisms, however, varies between species. For example, rat and mouse myocytes rely primarily on Ca++ reuptake by the SR (i.e., the SR accounting for 92% of Ca++ transport).
|
| REGULATION OF THE FORCE OF CONTRACTION
|
| Because the heart represents an electrical syncytium, with all of the cardiac muscle cells contracting during a single beat, it is not possible to increase the force of contraction by recruiting more muscle cells. Moreover, tetany of the heart would be lethal because it would defeat the critical pumping action of the heart. The heart has therefore developed alternative strategies to increase the force of contraction. It should be noted that the long action potential found in cardiac muscle, which is due to activation of the voltage-gated L-type Ca++ channel, results in a long refractory period, which in turn prevents tetany. Modulation of Ca++ influx through L-type Ca++ channels during an action potential, however, provides the heart with a mechanism to alter cytosolic [Ca++] and hence the force of contraction.
|
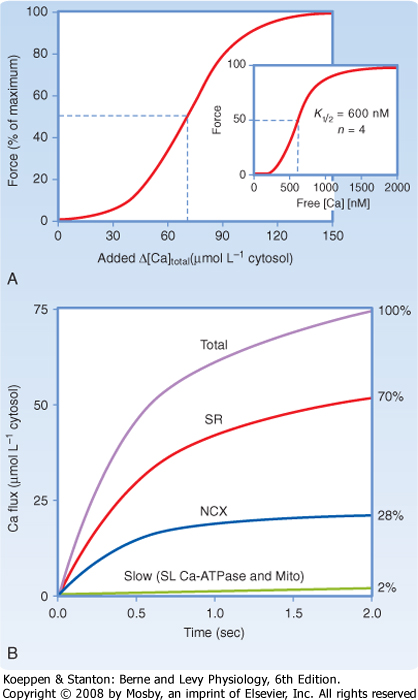
|
| Figure 13-4 Half-maximal force of contraction of cardiac muscles requires a rise in cytosolic free [Ca++] to approximately 600 nM (inset to panel A). Because of the high Ca++ buffering capacity of cytosolic proteins (such as parvalbumin and troponin C), this rise in free Ca++ requires an increase in total cytosolic [Ca++] of about 70 μM (panel A). Relaxation of the heart occurs by reducing cytosolic free [Ca++], with Ca++ sequestration by the SR accounting for the majority of the decrease in cytosolic [Ca++] (≈70%; panel B). Some Ca++ extrusion occurs through the 3Na+-1Ca++ antiporter (≈28%), with very little Ca++ extrusion by the sarcolemmal Ca++ pump (<2%). NCX, sodium-calcium exchanger. (Redrawn from Bers DM: Nature 415:198-205, 2002.) |
| page 261 | | | page 262 |
| A simple means of modulating the force of contraction of cardiac muscle cells in vitro is to vary extracellular [Ca++]. As noted previously, contraction of the heart requires extracellular Ca++. Decreasing extracellular [Ca++] from a normal range of 1 to 2 mM to 0.5 mM, for example, reduces the force of the contraction. This reduction in force of contraction is not associated with a change in the duration of the contraction because the kinetics of Ca++ sequestration by the SR and Ca++ extrusion has not been modified. Although
this approach of varying extracellular [Ca++] to alter the force of contraction is demonstrable in vitro, it is not a common means of modulating the force of cardiac contraction in vivo.
|
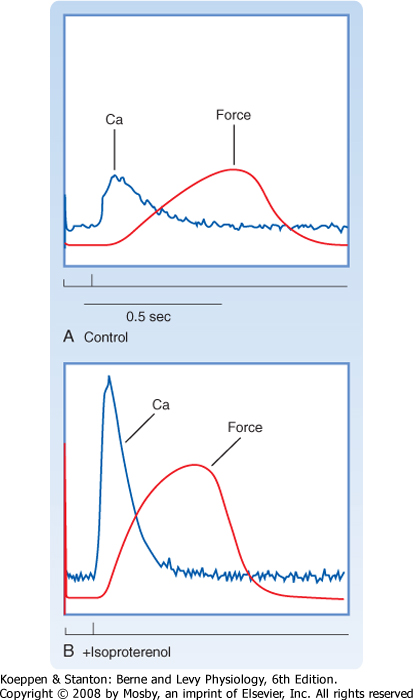
|
| Figure 13-5 Stimulation of β-adrenergic receptors in the heart increases the force of contraction. Electrical stimulation of myocardium results in a transient rise in intracellular [Ca++] and production of force (A). Isoproterenol (a β-adrenergic receptor agonist) increases the amplitude of the intracellular Ca++ transient and hence the amount of force generated (B). |
| In vivo, an increase in the size of the intracellular Ca++ transient and hence the force of contraction occurs in response to sympathetic stimulation (see later and also Chapter 18). Sympathetic stimulation often occurs during periods of excitement or fright and involves activation of β-adrenergic receptors on the heart by norepinephrine (released from nerve terminals in the heart) or epinephrine (released from the adrenal medulla into the bloodstream). As shown in Figure 13-5, the β-adrenergic agonist isoproterenol results in a dramatic increase in the size of the intracellular Ca++ transient and, consequently, a more forceful contraction. An increase in the force of contraction
is termed positive inotropy. Typically, there is also an increase in the rate of relaxation accompanying this β-adrenergic stimulation that results in a shorter contraction. The increase in the rate of muscle relaxation is termed positive lusitropy. The frequency of contractions of the heart also increases with β-adrenergic stimulation and is termed positive chronotropy. Thus, β-adrenergic stimulation of the heart produces stronger, briefer, and more frequent contractions.
|
| The sympathetic nervous system is stimulated when we become excited and is said to prepare the individual for "fight or flight." In the case of the heart, increased levels of the adrenal medullary hormone epinephrine or the sympathetic neurotransmitter norepinephrine activate β-adrenergic receptors on the cardiac muscle cells, which in turn activates adenylate cyclase, increases cAMP, and thus promotes cAMP-dependent phosphorylation of numerous proteins in cardiac muscle cells (Fig. 13-6).
|
| Both voltage-gated L-type Ca++ channels (responsible for the trigger Ca++) and a protein associated with SERCA, called phospholamban, are phosphorylated by cAMP-dependent protein kinase. The combined action of these phosphorylations increases the amount of Ca++ in the SR. Specifically, phosphorylation of the sarcolemmal Ca++ channel results in more trigger Ca++ entering the cell, and phosphorylation of phospholamban increases the activity of SERCA, thereby allowing the SR to accumulate more Ca++ before it is extruded by the 3Na+-1Ca++ antiporter and sarcolemmal Ca++ pump. The net result is that the SR releases more Ca++ into the cytosol during the next action potential, which promotes more actin-myosin interactions and hence greater force of contraction (Fig. 13-6). The increased activity of SERCA after sympathetic stimulation also results in a shortened contraction because of the rapid reaccumulation of Ca++ by the SR. This in turn allows the heart to increase its rate of relaxation. An additional consequence of sympathetic stimulation is an increase in heart rate through a direct effect on the pacemaker cells (see Chapter 18).
|
| The mechanisms underlying the response of the heart to β-adrenergic stimulation is complex and involves cAMP-dependent phosphorylation of several proteins. An A kinase adapter protein (AKAP) has been shown to be closely associated with the L-type Ca++ channel in the heart, thereby positioning cAMP-dependent protein kinase close to the channel and facilitating cAMP-dependent phosphorylation of this channel during sympathetic stimulation. How these cAMP-dependent phosphorylations increase the amplitude of the intracellular Ca++ transient and in so doing result in a more forceful, briefer cardiac contraction is discussed in general terms later (see also Chapter 18). |
| page 262 | | | page 263 |
| Figure 13-6 Sympathetic stimulation of the heart results in an increase in cytosolic cAMP and hence phosphorylation of several proteins by protein kinase A (PKA). An A kinase adapter protein (AKAP) adjacent to the L-type Ca++ channel facilitates phosphorylation of this channel and possibly nearby SR Ca++ channels (RyR). Other proteins phosphorylated by PKA include phospholamban (PLB) and troponin I. Muscarinic agonists (e.g., acetylcholine [ACh]), on the other hand, inhibit this sympathetic cascade by inhibiting the production of cAMP by adenylate cyclase (AC). β-AR, β-adrenergic receptor. (Redrawn from Bers DM: Nature 415:198, 2002.) |
| Mutations in the cardiac ryanodine receptor (RyR2) have been associated with cardiac arrhythmias. Specifically, catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited autosomal dominant disease that is typically manifested during childhood as an exercise-induced tachycardia that can progress to arrhythmias during exercise (or stress) and result in sudden death. Approximately 40% of patients with CPVT exhibit a defect in RyR2 that has been associated with increased release of Ca++ from the SR. The mutation in RyR2 may involve substitution of a highly conserved amino acid, which differs from malignant hyperthermia, in which splicing errors or deletions within the RYR have been reported. It is hypothesized that during periods of exercise or stress, increased levels of intracellular Ca++ (because of the combined effects of β-adrenergic stimulation and increased activity of the mutated RyR2) promote the development of delayed afterdepolarizations (DADs) and hence arrhythmias. Elevation of intracellular [Ca++] during diastole is thought to promote the development of DADs through activation of the 3Na+-1Ca++ antiporter, wherein Ca++ extrusion during diastole results in a net inward current sufficient to depolarize the cell to the threshold for an action potential. Treatment of CPVT patients involves antiadrenergic therapy (using β-adrenergic antagonists) or (for unresponsive patients) an implanted defibrillator. |
|
| Stretching the heart increases the force of contraction both in vivo and in vitro and is an intrinsic mechanism for regulating contractile force. This contrasts with skeletal muscle, which typically exhibits maximal tension at resting length. Stretching of the heart in vivo occurs during times of increased venous return of blood to the heart (e.g., during exercise or when the heart rate is slowed, or both). The Frank-Starling law of the heart refers to this ability of the heart to increase its force of contraction when stretched, which occurs at times of increased venous return (Fig. 13-7; also see Chapter 16).
|
| The importance of this mechanism is that it helps the heart pump whatever volume of blood it receives. Thus, when the heart receives a lot of blood, the ventricles are stretched and the force of contraction is increased, thereby ensuring ejection of this extra volume of blood. It should be noted that stretching cardiac muscle also increases passive tension, which helps prevent overstretching of the heart. This passive resistance in the heart is greater than that in skeletal muscle and is attributed to both extracellular matrix (connective tissue) and intracellular elastic proteins (titin).
|
| page 263 | | | page 264 |
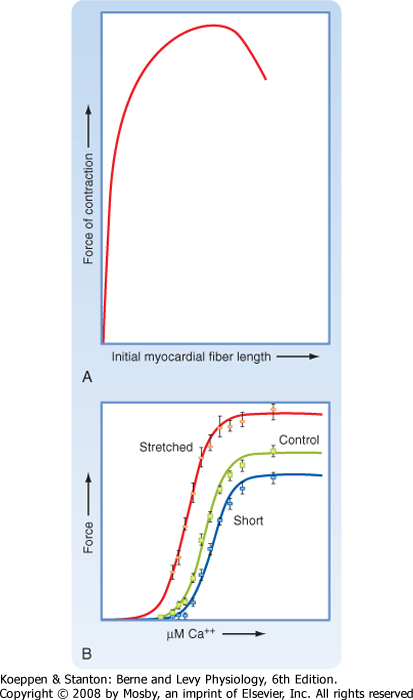
| Figure 13-7 Stretching the heart increases the force of contraction (A). This is attributable to both an increase in the maximal force of contraction and an increase in the sensitivity of contraction to Ca++ (B) and reflects an intrinsic regulatory process referred to as the Frank-Starling law of the heart. |
| The mechanism underlying this stretch-induced increase in force of contraction appears to involve a change in both sensitivity to Ca++ and the level of actin-myosin interactions. The change in the level of actin-myosin interactions is shown in Figure 13-7, B. When compared with control cardiac muscle, stretched cardiac muscle exhibits an increased force of contraction at saturating [Ca++]. Likewise, shortening cardiac muscle (by precontracting the muscle) results in a less forceful contraction at saturating [Ca++] than with either control or stretched cardiac muscle. The mechanism underlying this length-dependent increase in force does not appear to be due to differences in the overlap of thick and thin filaments. Instead, evidence suggests that stretch reduces the space between thick and thin filaments (i.e., interfilament spacing), and this is associated with the ability of more myosin
molecules to interact with actin (increasing the force at saturating [Ca++]). The intracellular elastic protein titin has been implicated in this length-dependent increase in force because it binds both actin and myosin and might pull the actin and myosin filaments closer together when the muscle/titin is stretched (Fig. 13-8). Consistent with the latter hypothesis, in vitro experiments involving partial proteolysis of titin resulted in attenuation of the length-dependent increase in force.
|
| Stretch also increases sensitivity to Ca++ and the level of actin-myosin interactions in cardiac muscle (Fig. 13-7, B). Stretched cardiac muscle requires a lower level of Ca++ to produce a half-maximal force of contraction than control/unstretched cardiac muscle does. Moreover, precontracted cardiac muscle requires a higher level of Ca++ to produce a half-maximal force of contraction than control/unstretched cardiac muscle does. The mechanism or mechanisms underlying this stretch-induced increase in the sensitivity of cardiac actin-myosin interactions to Ca++ are unknown but probably also involve a stretch-induced decrease in spacing between actin and myosin filaments, perhaps involving titin. Interestingly, skeletal muscle does not exhibit this stretch-dependent change in sensitivity to Ca++, although it does contain titin. This difference between muscle types may reflect the expression of different titin isoforms or other proteins (e.g., myosin, troponins, and tropomyosin).
|
| CARDIAC MUSCLE METABOLISM
|
| As in skeletal muscle, myosin uses the energy in ATP to generate force, so the ATP pool, which is small, must be continually replenished. Typically, this replenishment of ATP pools is accomplished by aerobic metabolism, including the oxidation of fats and carbohydrates. During times of ischemia, the creatine phosphate pool, which converts ADP to ATP, may decrease. As in skeletal muscle, the creatine phosphate pool is small.
|
| When cardiac muscle is completely deprived of O2 because of occlusion of a coronary vessel (i.e., stopped-flow ischemia), contractions quickly cease (within 30 seconds). This is not due to depletion of either ATP or creatine phosphate because these levels decline more slowly. Even after 10 minutes of stopped-flow ischemia, when creatine phosphate levels are near zero and only 20% of the ATP remains, reperfusion can restore these energy stores, as well as contractile ability. However, prolonging the stopped-flow ischemia for 20 minutes results in further drops in ATP such that reperfusion has considerably less effect with only limited restoration of ATP and creatine phosphate levels or contractile activity.
|
| CARDIAC MUSCLE HYPERTROPHY
|
| Exercise such as endurance running can increase the size of the heart as a result of hypertrophy of individual cardiac muscle cells. Concomitant with this enlarged "athlete's heart" is improved cardiac performance, as assessed by an increase in stroke volume, increased oxygen consumption, and preserved relaxation. Thus, the "athlete's heart" represents an example of "physiological hypertrophy," with beneficial contractile effects.
|
| By contrast, if exposed to chronic pressure overload, the heart may undergo either concentric left ventricular hypertrophy or dilated left ventricular hypertrophy, with impaired functional consequences. Details regarding the morphological, functional, and mechanistic differences between these various types of hypertrophy can be found elsewhere in this textbook (see Chapter 18).
|
| page 264 | | | page 265 |
| Figure 13-8 Titin may contribute to the ability of stretch to increase the force of contraction of the heart. Titin binds to both myosin and actin such that stretch of the cardiac muscle may bring the actin filament closer to the myosin head and thus increase the number of myosin heads that interact with actin at a given intracellular [Ca++]. (Redrawn from Moss RL, Fitzsimons DP: Circ Res 90:11-13, 2002.) |
| Concentric hypertrophy is characterized by thickening of the left ventricular wall and represents a compensatory hypertrophy to the increased load. Dilated hypertrophy is characterized by increased ventricular volume (end-diastolic volume). Both concentric/compensatory left ventricular hypertrophy and dilated left ventricular hypertrophy have been shown to exhibit decreased contractile response to β-adrenergic stimulation, thus limiting the contractile reserve. In dilated left ventricular hypertrophy, normal contractile function, along with the Frank-Starling response, may also be impaired.
|
| The cellular and molecular mechanism or mechanisms underlying the development of cardiac hypertrophy are not clear, although an elevation in intracellular [Ca++] has been implicated.
|
| The link or links between cardiac hypertrophy, decreased cardiac performance, and impaired β-adrenergic response during chronic pressure overload are unclear. Decreased cardiac performance has been attributed to dysregulation of intracellular [Ca++]. Alterations in the level, activity, and phosphorylation status of a variety of proteins, including L-type Ca++ channels, phospholamban, SERCA, and RYR, have all been implicated in the Ca++ dysregulation associated with a failing heart (pathological hypertrophy).
|
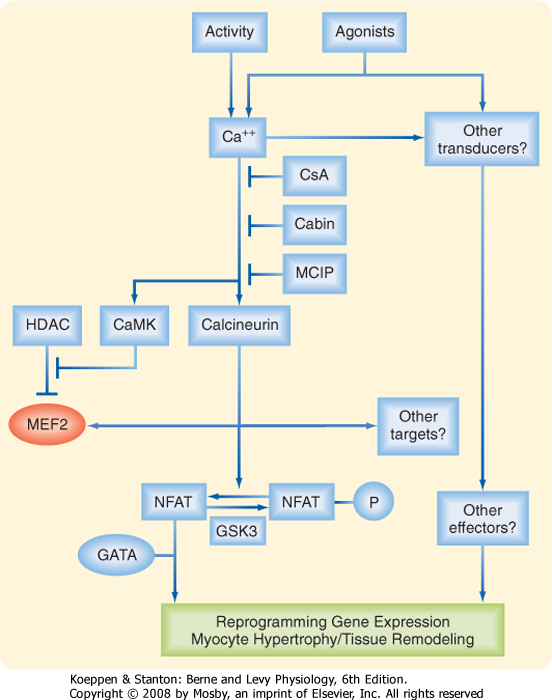
| A modest elevation in intracellular [Ca++] (as a result of increased contractile activity), for example, has been proposed to activate a Ca++-calmodulin-dependent protein phosphatase (calcineurin) that can dephosphorylate the transcription factor NFAT (nuclear factor of activated T cells), thereby facilitating translocation of NFAT to the nucleus and ultimately promoting protein synthesis and thus hypertrophy (Fig. 13-9). Activation of Ca++-calmodulin-dependent protein kinase has also been implicated in activation of the transcription factor MEF2 (Myocyte Enhancer Factor 2) by promoting the dissociation (nuclear export) of an inhibitor of MEF2 (viz., histone deacetylase [HDAC]). |
|
| The impaired β-adrenergic response of cardiac muscle after chronic pressure overload involves, at least in part, a decrease in β-adrenergic receptors because of internalization. Both phosphatidylinositol-3-kinase (PI3K) and β-adrenergic receptor kinase 1 have been implicated in the internalization of β-adrenergic receptors.
|
| page 265 | | | page 266 |
| Figure 13-9 Calcium-dependent activation of calcineurin and calmodulin-dependent protein kinase have been implicated in the development of cardiac hypertrophy and involve activation of the transcription factors NFAT, GATA, and MEF2. Cabin, calcineurim binding protein/inhibitor; CaMK, Ca++/calmodulin dependent protein kinase; CsA, cyclosporin; GATA, transcription factor binding to DNA sequence GATA; GSK3, glycogen synthase kinase 3; HDAC, histone deacetylase; MCIP, modulatory calcineurin interacting protein; MEF2, myocyte enhancer factor 2; NFAT, nuclear factor of activated T-cells. (Redrawn from Olson EN, Williams RS: Cell 101:689-692, 2000.) |
| High blood pressure, defects in heart valves, and weakened ventricular walls secondary to myocardial infarction can all lead to heart failure, a leading cause of death. Heart failure may be seen with thickening of the walls of the ventricle or with dilation (i.e., increased volume) of the ventricles. |
| Studies suggest that dilated cardiomyopathy can be prevented in an animal model by down-regulating a protein called phospholamban. The mechanism underlying this preventive effect of phospholamban down-regulation is thought to involve an increase in SR Ca++ uptake activity because phospholamban typically inhibits SERCA. Increased activity of SERCA would facilitate relaxation of the heart as a result of rapid Ca++ uptake by the SR. In addition, the force of contraction is increased because more Ca++ is available for release. Increased Ca++ uptake by the SR may also decrease activation of the Ca++-dependent phosphatases that have been implicated in the development of cardiac hypertrophy. |
|
| Finally, there is evidence that cardiac hypertrophy can be dissociated from some functional impairments. Intermittent aortic constrictions, for example, result in decreased β-adrenergic signaling, decreased capillary density, and decreased SERCA2 levels, without evidence of hypertrophy. Activation of PI3K appears to be involved in this response.
|
| page 266 | | | page 267 |
- Cardiac muscle is an involuntary, striated muscle. Cardiac muscle cells are relatively small (10 μm × 100 μm) and form an electrical syncytium with tight electrical and mechanical connections between adjacent cardiac muscle cells. Action potentials are initiated in the sinoatrial node and spread quickly throughout the heart to allow synchronous contraction, a feature important for the pumping action of the heart.
- Contraction of cardiac muscle involves the Ca++-dependent interaction of actin and myosin filaments, as in skeletal muscle. However, unlike skeletal muscle, an influx of extracellular Ca++ is required. Specifically, the influx of Ca++ during an action potential triggers release of Ca++ from the SR, which then promotes actin-myosin interaction and contraction.
- Relaxation of cardiac muscle involves reaccumulation of Ca++ by the SR and extrusion of Ca++ from the cell via the 3Na+-1Ca++ antiporter and the sarcolemmal Ca++ pump.
- The force of contraction of cardiac muscle is increased by stretch (Frank-Starling law of the heart) and by sympathetic stimulation. This differs from skeletal muscle, which increases force by recruiting more muscle fibers or by tetany.
- Hypertrophy of the heart can occur in response to exercise, chronic pressure overload, or genetic mutations. The cardiac hypertrophy resulting from exercise is typically beneficial, with improved cardiac performance, increased oxygen consumption, and normal relaxation. Chronic pressure overload, on the other hand, can result in cardiac hypertrophy that is initially associated with a decreased β-adrenergic response but may progress to dilated cardiac hypertrophy characterized by decreased contractile ability. Genetic mutations resulting in cardiac hypertrophy include familial hypertrophic cardiomyopathy, in which a mutation in a single intracellular protein may alter contractile function and promote a hypertrophic response.
|
|
|