| Nonstriated, or smooth, muscle cells are a major component of hollow organs such as the alimentary canal, airways, vasculature, and urogenital tract. Contraction of smooth muscle serves to alter the dimensions of the organ, which may result in either propelling the contents of the organ (as in peristalsis of the intestine) or increasing the resistance to flow (as in vasoconstriction). The basic mechanism underlying contraction of smooth muscle involves an interaction of myosin with actin (as in striated muscle), although there are some important differences. Specifically, contraction of smooth muscle is thick filament regulated and requires an alteration in myosin before it can interact with actin, whereas contraction of striated muscle is thin filament regulated and requires movement of the troponin-tropomyosin complex on the actin filament before myosin can bind to actin. Smooth muscle can contract in response to either electrical or hormonal signals and exhibits the ability to remain contracted for extended periods at low levels of energy consumption, which is important for functions such as maintaining vascular tone and hence blood pressure. An additional feature of smooth muscle (termed "length adaptation") facilitates contraction of smooth muscle over a broad range of lengths, which may be important for emptying a hollow organ at various levels of filling. Thus, regulation of contraction of smooth muscle is complex, sometimes involving multiple intracellular signaling cascades. In the present chapter, effort is made to identify mechanisms underlying this diverse regulation of smooth muscle contraction and, when appropriate, compare these regulatory mechanisms with those observed in striated muscle. Alterations in smooth muscle function/regulation that have been implicated in various pathological conditions are also discussed.
|
| OVERVIEW OF SMOOTH MUSCLE
|
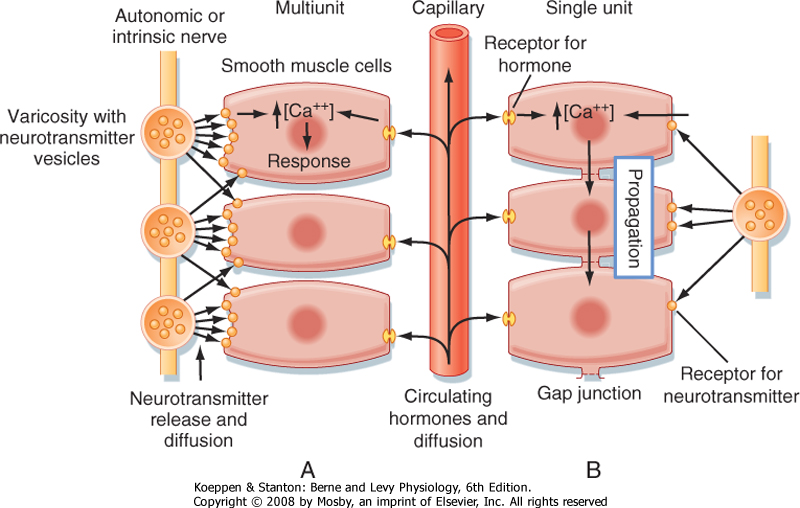
| Smooth muscle has been subdivided into two groups: single unit and multiunit. In single-unit smooth muscle, the smooth muscle cells are electrically coupled such that electrical stimulation of one cell is followed by stimulation of adjacent smooth muscle cells. This results in a wave of contraction, as in peristalsis. Moreover, this wave of electrical activity, and hence contraction, in single-unit smooth muscle may be initiated by a pacemaker cell (i.e., a smooth muscle cell that exhibits spontaneous depolarization). In contrast, multiunit smooth muscle cells are not electrically coupled, so stimulation of one cell does not necessarily result in activation of adjacent smooth muscle cells. Examples of multiunit smooth muscle include the vas deferens of the male genital tract and the iris of the eye. Smooth muscle, however, is even more diverse, with the single-unit and multiunit classifications representing ends of a spectrum. Moreover, the terms single-unit and multiunit represent an oversimplification because most smooth muscles are modulated by a combination of neural elements, with at least some degree of cell-to-cell coupling, and locally produced activators or inhibitors, which also promote a somewhat coordinated response of smooth muscles.
|
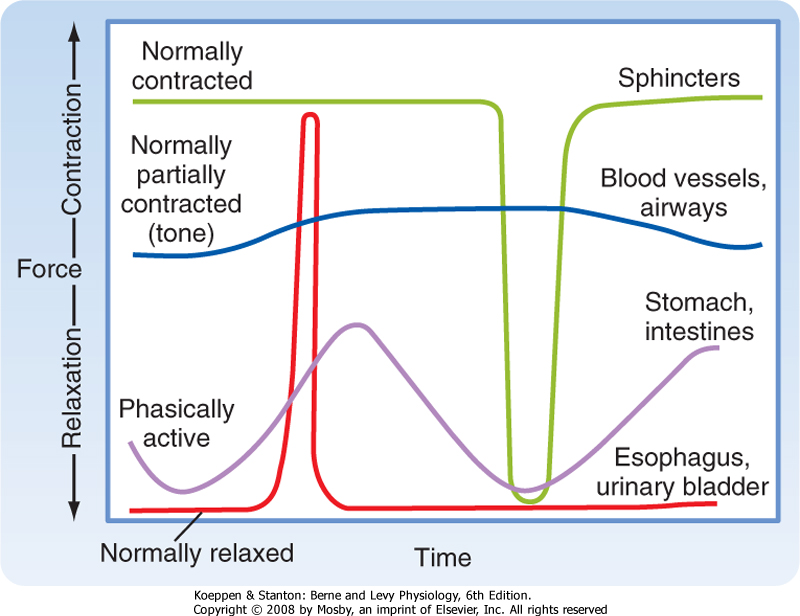
| A second consideration when discussing types of smooth muscle is the activity pattern (Fig. 14-1). In some organs, the smooth muscle cells contract rhythmically or intermittently, whereas in other organs, the smooth muscle cells are continuously active and maintain a level of "tone." Smooth muscle exhibiting rhythmic or intermittent activity is termed phasic smooth muscle and includes smooth muscles in the walls of the gastrointestinal and urogenital tracts. Such phasic smooth muscle corresponds to the single-unit category described earlier because the smooth muscle cells contract in response to action potentials that propagate from cell to cell. Smooth muscle that is continuously active, on the other hand, is termed tonic smooth muscle. Vascular smooth muscle, respiratory smooth muscle, and some sphincters are continuously active. The continuous partial activation of tonic smooth muscle is not associated with action potentials, although it is proportional to membrane potential. Tonic smooth muscle would thus correspond to the multiunit smooth muscle described earlier. Phasic and tonic contractions of smooth muscle result from interactions of actin and myosin filaments, although as discussed later in this chapter, there is a change in cross-bridge cycling kinetics during tonic contraction such that the smooth muscle can maintain force at low energy cost.
|
| STRUCTURE OF SMOOTH MUSCLE CELLS
|
| page 268 |  | | page 269 |
| Figure 14-1 Some contractile activity patterns exhibited by smooth muscles. Tonic smooth muscles are normally contracted and generate a variable steady-state force. Examples are sphincters, blood vessels, and airways. Phasic smooth muscles commonly exhibit rhythmic contractions (e.g., peristalsis in the gastrointestinal tract) but may contract intermittently during physiological activities under voluntary control (e.g., voiding of urine and swallowing). |
| Smooth muscle cells typically form layers around hollow organs (Fig. 14-2). Blood vessels and airways exhibit a simple tubular structure in which the smooth muscle cells are arranged circumferentially, so contraction
reduces the diameter of the tube. This contraction increases resistance to the flow of blood or air but has little effect on the length of the organ. Smooth muscle cell organization is more complex in the gastrointestinal tract. Layers of smooth muscle in both circumferential and longitudinal orientations provide the mechanical action for mixing food and also propelling the luminal contents from the mouth to the anus. Coordination between these layers
depends on a complex system of autonomic nerves linked by plexuses. These plexuses are located between the two muscle layers. The smooth muscle in the walls of saccular structures, such as the urinary bladder or rectum, allows the organ to increase in size with the accumulation of urine or feces. The varied arrangement of cells in the walls of these organs contributes to their ability to reduce internal volume to almost zero during urination or defecation. Smooth muscle cells in hollow organs occur in a spectrum of forms, depending on their function and mechanical loads.
|
| In all hollow organs, the smooth muscle is separated from the contents of the organ by other cellular elements, which may be as simple as vascular endothelium or as complex as the mucosa of the digestive tract. The walls of hollow organs also contain large amounts of connective tissue that bear an increasing share of the wall stress as organ volume increases.
|
| The following sections describe the structural components that enable smooth muscle to set or alter hollow organ volume. These components include contractile and regulatory proteins, force-transmitting systems such as the cytoskeleton, linkages between cells and to the extracellular matrix, and membrane systems that transduce extracellular signals into changes in myoplasmic [Ca++].
|
| Figure 14-2 Scanning electron micrographs of smooth muscle. A, Muscular arteriole with fusiform smooth muscle cells in a circular orientation (bar, 20 μm). B, Superimposed images of circular (below) and longitudinal (above) layers of intestinal smooth muscle sandwiching neural components of the myenteric plexus (asterisk) (bar, 50 mm). C, Rectangular smooth muscle cells with thin projections to adjacent cells in a small testicular duct (bar, 5 μm). (From Motta PM [ed]: Ultrastructure of Smooth Muscle. Norwell, MA, Kluwer Academic, 1990.) |
| page 269 | | | page 270 |
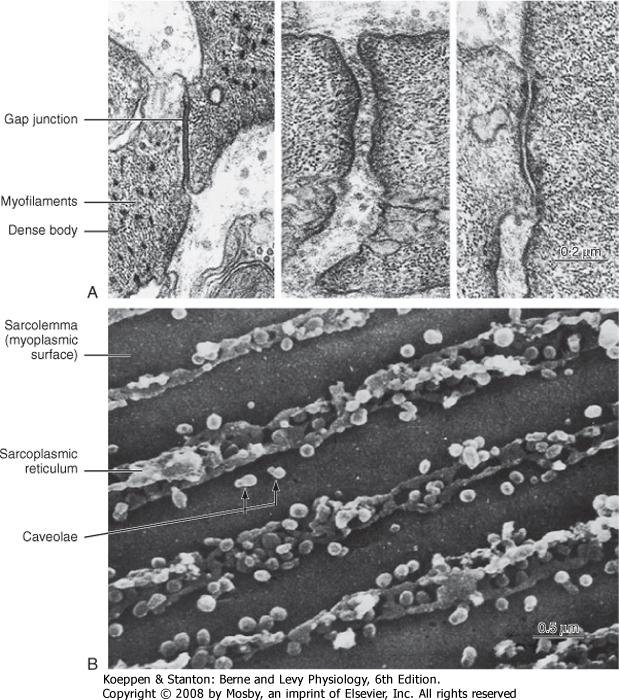
| Figure 14-3 Junctions and membranes in smooth muscle. A, Transmission electron micrograph of junctions between intestinal smooth muscle cells. B, Scanning electron micrograph of the inner surface of the sarcolemma of an intestinal smooth muscle cell. Longitudinal rows of caveolae project into the myoplasm (small, light-colored spheres), surrounded by darker elements of the tubular sarcoplasmic reticulum. The attachments of thin filaments to the sarcolemma between the rows of membrane elements were removed during preparation of the specimen. (From Motta PM [ed]: Ultrastructure of Smooth Muscle, Norwell, MA, Kluwer Academic, 1990.) |
| A variety of specialized contact exists between smooth muscle cells. Such contact allows mechanical linkage and communication between the cells (Fig. 14-3). In contrast to skeletal muscle cells, which are normally attached at either end to a tendon, smooth (and cardiac) muscle cells are connected to each other. Because smooth muscle cells are anatomically
arranged in series, they not only must be mechanically linked but must also be activated simultaneously and to the same degree. This mechanical and functional linkage is crucial to smooth muscle function. If such linkage did not exist, contraction in one region would simply stretch another region without a substantial decrease in radius or increase in pressure. The mechanical connections are provided by attachments to sheaths of connective tissue and by specific junctions between muscle cells.
|
| Several types of junctions are found in smooth muscle (Fig. 14-4). Functional linkage of the cells is provided by gap junctions. Gap junctions form low-resistance pathways between cells (see Chapter 2). They also allow chemical communication by diffusion of low-molecular-weight compounds. In certain tissues, such as the outer longitudinal layer of smooth muscle in the intestine, large numbers of such junctions exist. Action potentials are readily propagated from cell to cell through such tissues.
|
| Adherens junctions (also called dense plaques or attachment plaques) provide mechanical linkage between smooth muscle cells. As depicted in Figure 14-4, the adherens junction appears as thickened regions of opposing cell membranes that are separated by a small gap (≈60 nm) containing dense granular material. Thin filaments extend into the adherens junction to allow the contractile force generated in one smooth muscle cell to be transmitted to adjacent smooth muscle cells.
|
| page 270 | | | page 271 |
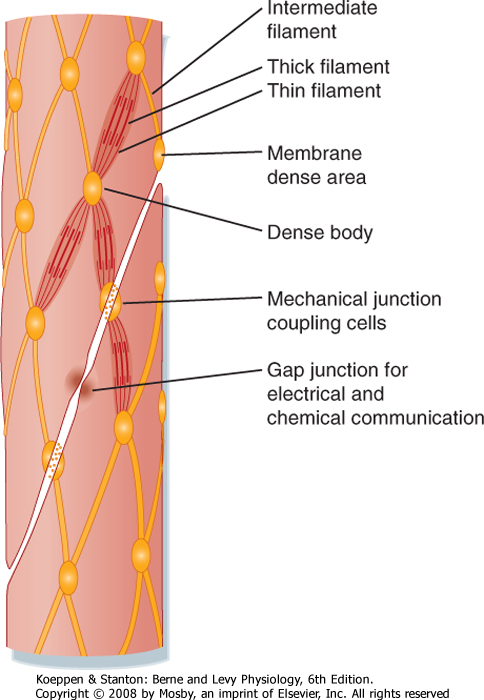
| Figure 14-4 Apparent organization of cell-to-cell contacts, cytoskeleton, and myofilaments in smooth muscle cells. Small contractile elements functionally equivalent to a sarcomere underlie the similarities in mechanics between smooth and skeletal muscle. Linkages consisting of specialized junctions or interstitial fibrillar material functionally couple the contractile apparatus of adjacent cells. |
| Figure 14-3 also shows the presence of caveolae, which represent invaginations of the smooth muscle membrane (analogous to T tubules in striated muscle). The sarcoplasmic reticulum (SR) extends throughout the smooth muscle cell, although as depicted in Figure 14-3, there are junctional regions of the SR where it abuts regions of the sarcolemma or caveolae, or both. As discussed in a subsequent section, these subsarcolemmal regions of the SR play an important role in the regulation of intracellular [Ca++] and hence smooth muscle tone.
|
| Embryonic smooth muscle cells do not fuse, and each differentiated cell has a single, centrally located nucleus (Fig. 14-5). Though dwarfed by skeletal muscle cells, smooth muscle cells are nevertheless quite large (typically 40 to 600 μm long). These cells are 2 to 10 μm in diameter in the region of the nucleus, and most taper toward their ends. Contracting cells become quite distorted as a result of the force exerted on the cell by attachments to other cells or to the extracellular matrix, and cross sections of these cells are often very irregular.
|
| Smooth muscle cells lack T tubules, the invaginations of the skeletal muscle sarcolemma that provide electrical links to the SR. However, the sarcolemma of smooth muscle has longitudinal rows of tiny sac-like in-pocketings called caveolae (Figs. 14-3 and 14-5). Caveolae increase the surface-to-volume ratio of the cells and are often closely opposed to the underlying SR. A gap of approximately 15 nm has been observed between the caveolae and the underlying SR, comparable to the gap between the T tubules and terminal SR in skeletal muscle. Moreover, "Ca++ sparks" and a variety of Ca++-handling proteins have been observed in the vicinity of caveolae, thus raising the possibility that the caveolae and the underlying SR may contribute to the regulation of intracellular [Ca++] in smooth muscle. The voltage-gated L-type Ca++ channel and the 3Na+-1Ca++ antiporter, for example, are associated with caveolae. The proteins caveolin and cholesterol are both critical for the formation of caveolae, and it is hypothesized that the caveolae reflect a specialized region of the sarcolemma that may also contain various signaling molecules in addition to the Ca++ signaling mentioned earlier.
|
| Smooth muscle also has an intracellular membrane network of SR that serves as an intracellular reservoir for Ca++ (Figs. 14-3 and 14-5). Calcium can be released from the SR into the myoplasm when stimulatory neurotransmitters, hormones, or drugs bind to receptors on the sarcolemma. Importantly, intracellular Ca++ channels in the SR of smooth muscle include the ryanodine receptor (RYR), which is similar to that found in skeletal muscle SR, and the inositol 1,4,5-trisphosphate (InsP3)-gated Ca++ channel. The RYR is typically activated by a rise in intracellular [Ca++] (i.e., Ca++-induced release of Ca++ in response to an influx of Ca++ through the sarcolemma). The InsP3-gated Ca++ channel is activated by InsP3, which is produced when a hormone or hormones bind to various Ca++-mobilizing receptors on the sarcolemma. Intracellular [Ca++] is lowered through the action of an SR Ca++-ATPase (SERCA) and extrusion of Ca++ from the cell via a 3Na+-1Ca++ antiporter and a sarcolemmal Ca++-ATPase. The amount of SR in smooth muscle cells varies from 2% to 6% of cell volume and approximates that of skeletal muscle. As mentioned earlier, chemical signals such as InsP3 or a localized increase in intracellular [Ca++] (e.g., within the gap between the caveolae and SR) functionally link the sarcolemma and the SR.
|
| Smooth muscle cells contain a prominent rough endoplasmic reticulum and Golgi apparatus, which are located centrally at each end of the nucleus. These structures reflect significant protein synthetic and secretory functions. The scattered mitochondria (Fig. 14-5) are sufficient for oxidative phosphorylation to generate the increased ATP consumed during contraction.
|
| page 271 | | | page 272 |
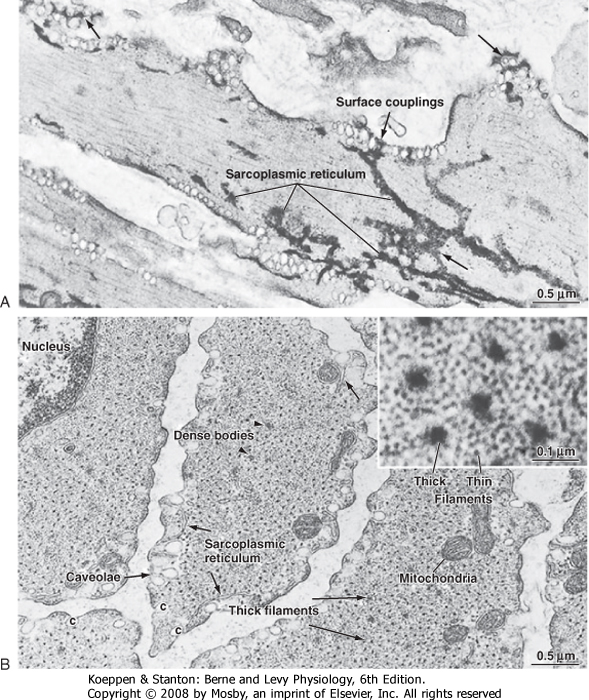
| Figure 14-5 A, Longitudinal view of a pulmonary artery smooth muscle cell. The sarcoplasmic reticulum is stained with osmium ferricyanide and appears to form a continuous network throughout the cell consisting of tubules, fenestrated sheets (long arrows), and surface couplings at the cell membrane (short arrows). B, Transverse section of a bundle of venous smooth muscle cells illustrating the regular spacing of thick filaments (long arrows) and the relatively large number of surrounding thin (actin) filaments (inset). Dense bodies (arrowheads) are sites of attachment for the thin actin filaments and equivalent to the Z lines of striated muscles. Elements of sarcoplasmic reticulum (short arrows) occur at the periphery of these cells. (From Somlyo AP, Somlyo AV: Smooth muscle structure and function. In Fozzard HA et al [eds]: The Heart and Cardiovascular System, 2nd ed. New York, Raven Press, 1992.) |
| The thick and thin filaments of smooth muscle cells are about 10,000 times longer than their diameter and are tightly packed. Therefore, the probability of observing an intact filament by electron microscopy is extremely low. In contrast to skeletal muscle, which contains a transverse alignment of thick and thin filaments that results in striations, the contractile filaments in smooth muscle are not in uniform transverse alignment, and thus smooth muscle has no striations.
The lack of striations in smooth muscle does not imply a lack of order. The thick and thin filaments are organized in contractile units that are analogous to sarcomeres.
|
| page 272 | | | page 273 |
| The thin filaments of smooth muscle have an actin and tropomyosin composition and structure similar to that in skeletal muscle. However, the cellular content of actin and tropomyosin in smooth muscle is about twice that of striated muscle. Smooth muscle lacks troponin and nebulin but contains two proteins not found in striated muscle: caldesmon and calponin. The precise roles of these proteins are unknown, but they do not appear to be fundamental to cross-bridge cycling. It has been suggested that calponin may inhibit the binding of unphosphorylated myosin to actin. Most of the myoplasm is filled with thin filaments that are roughly aligned along the long axis of the cell. The myosin content of smooth muscle is only a fourth that of striated muscle. Small groups of three to five thick filaments are aligned and surrounded by many thin filaments. These groups of thick filaments with interdigitating thin filaments are connected to
dense bodies or areas (Figs. 14-4 and 14-5) and represent the equivalent of the sarcomere. To maintain alignment of the contractile apparatus along the long axis of the cell, the thick and thin filaments of some smooth muscles do not appear to circumvent the centrally located nucleus but instead may connect to (or near) the nucleus. The contractile apparatus of adjacent cells is mechanically coupled by the links between membrane-dense areas (Fig. 14-4).
|
| The cytoskeleton in smooth muscle cells serves as an attachment point for the thin filaments and permits transmission of force to the ends of the cell. In contrast to skeletal muscle, the contractile apparatus in smooth muscle is not organized into myofibrils, and Z lines are lacking. The functional equivalents of the Z lines in smooth muscle cells are ellipsoidal dense bodies in the myoplasm and dense areas that form bands along the sarcolemma (Figs. 14-3 to 14-5). These structures serve as attachment points for the thin filaments and contain α-actinin, a protein also found in the Z lines of striated muscle. Intermediate filaments with diameters between those of thin filaments (7 nm) and thick filaments (15 nm) are prominent in smooth muscle. These filaments link the dense bodies and areas into a cytoskeletal network (Fig. 14-4). The intermediate filaments consist of protein polymers of desmin or vimentin.
|
| CONTROL OF SMOOTH MUSCLE ACTIVITY
|
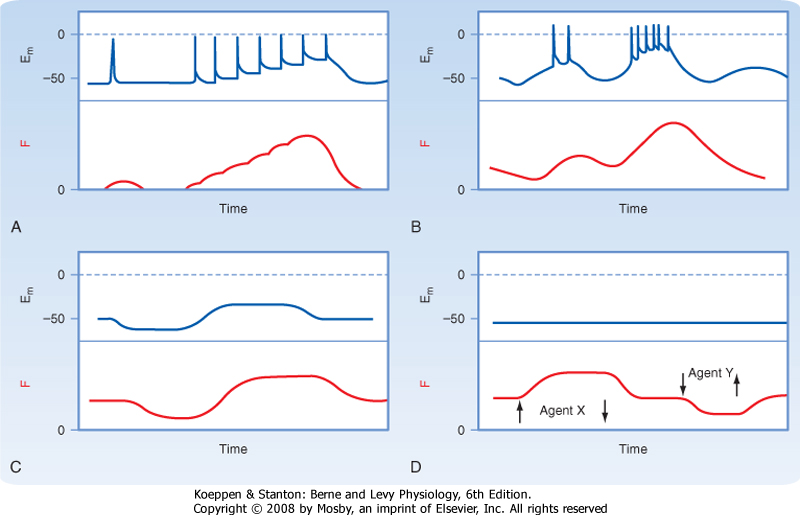
| The contractile activity of smooth muscle can be controlled by numerous factors, including hormones, autonomic nerves, pacemaker activity, and a variety of drugs. Like skeletal or cardiac muscle, contraction of smooth muscle is dependent on Ca++, and the agents just listed induce smooth muscle contraction by increasing intracellular [Ca++]. However, in contrast to skeletal or cardiac muscle, action potentials in smooth muscle are highly variable and not always needed to initiate contraction. Moreover, several agents can increase intracellular [Ca++] and hence contract smooth muscle without changing the membrane potential. Figure 14-6 shows various types of action potentials in smooth muscle and the corresponding changes in force. An action potential in smooth muscle can be associated with a slow twitch-like response, and the twitch forces can summate during periods of repetitive action potentials (i.e., similar to tetany in skeletal muscle). Such a pattern of activity is characteristic of single-unit smooth muscle in many viscera.
|
| Periodic oscillations in membrane potential can occur as a result of changes in the activity of Na+,K+-ATPase in the sarcolemma. These oscillations in membrane potential can trigger multiple action potentials in the cell. Alternatively, the contractile activity of smooth muscle may not be associated with the generation of action potentials or even a change in membrane potential. In many smooth muscles, the resting membrane potential is sufficiently depolarized (-60 to -40 mV) that a small decrease in membrane potential can significantly inhibit influx of Ca++ through voltage-gated Ca++ channels in the sarcolemma. By decreasing Ca++ influx, the force developed by smooth muscle decreases. Such a graded response to slight changes in the resting membrane potential is common in multiunit smooth muscles that maintain constant tension (e.g., vascular smooth muscle).
|
| Contraction of smooth muscle in response to an agent that does not produce a change in membrane potential is termed pharmacomechanical coupling and typically reflects the ability of the agent to increase the level of the intracellular second messenger InsP3. Other agents result in a decrease in tension, also without a change in membrane potential. These agents typically increase levels of the intracellular second messengers cGMP or cAMP. The molecular mechanisms by which InsP3, cGMP, cAMP, and Ca++ alter the contractile force of smooth muscle are presented later.
|
| Phosphorylation of a myosin light chain is required for the interaction of myosin with actin, and although Ca++-dependent phosphorylation plays a key role in this process, the level of myosin phosphorylation (and hence the degree of contraction) is dependent on the relative activities of both myosin light-chain kinase (MLCK, which promotes phosphorylation) and myosin phosphatase (MP, which promotes dephosphorylation). Several agonists/hormones increase the level of myosin light-chain phosphorylation by simultaneously activating MLCK through an increase in intracellular [Ca++] and inhibiting MP through a signaling cascade involving the monomeric G protein RhoA and its effector Rho kinase (ROK). Moreover, hyperactivity of this RhoA/ROK signaling cascade has been implicated in various pathological conditions such as hypertension and vasospasm (discussed later).
|
| INNERVATION OF SMOOTH MUSCLE
|
| Neural regulation of smooth muscle contraction depends on the type of innervation and neurotransmitters released, the proximity of the nerves to the muscle cells, and the type and distribution of the neurotransmitter receptors on the muscle cell membranes (Fig. 14-7). In general, smooth muscle is innervated by the autonomic nervous system. The smooth muscle in arteries is innervated primarily by sympathetic fibers, whereas the smooth muscle in other tissues can have both sympathetic and parasympathetic innervation. In the gastrointestinal tract, smooth muscle is innervated by nerve plexuses that make up the enteric nervous system. The smooth muscle cells of some tissues, such as the uterus, have no innervation.
|
| page 273 | | | page 274 |
|
| Figure 14-6 Relationships between membrane potential (Em) and generation of force (F) in different types of smooth muscle. A, Action potentials may be generated and lead to a twitch or larger summed mechanical responses. Action potentials are characteristic of single-unit smooth muscles (many viscera). Gap junctions permit the spread of action potentials throughout the tissue. B, Rhythmic activity produced by slow waves that trigger action potentials. The contractions are generally associated with a burst of action potentials. Slow oscillations in membrane potential usually reflect the activity of electrogenic pumps in the cell membrane. C, Tonic contractile activity may be related to the value of the membrane potential in the absence of action potentials. Graded changes in Em are common in multiunit smooth muscles (e.g., vascular), where action potentials are not generated and propagated from cell to cell. D, Pharmacomechanical coupling; changes in force produced by the addition or removal (arrows) of drugs or hormones that have no significant effect on membrane potential. |
| The neuromuscular junctions and neuromuscular transmission in smooth muscle are functionally comparable to that of skeletal muscle, but structurally less complex. The autonomic nerves that supply smooth muscle have a series of swollen areas, or varicosities, that are spaced at intervals along the axon. These varicosities contain vesicles for the neurotransmitter (Fig. 14-7). The postsynaptic membrane of smooth muscle exhibits little specialization when compared with that of skeletal muscle (see Chapter 6). The synaptic
cleft is typically about 80 to 120 nm wide but can be as narrow as 6 to 20 nm or even greater than 120 nm. In synapses in which a wide synaptic cleft is found, release of neurotransmitter can affect multiple smooth muscle cells. There are a large number of neurotransmitters that affect smooth muscle activity. A partial listing is provided in Table 14-1.
|
| REGULATION OF CONTRACTION
|
| The enteric nervous system controls many aspects of gastrointestinal function, including motility. Some children are born without enteric nerves in the distal portion of the colon. The absence of nerves is caused by mutant genes that disrupt the signals necessary for the embryonic nerves to migrate to the colon. In these children, normal motility of the colon does not occur and severe constipation results. This condition is called Hirschsprung's disease. It can be corrected by surgically removing the portion of the colon that does not contain enteric nerves. |
|
| Contraction of smooth muscle requires the phosphorylation of a myosin light chain. Typically, this phosphorylation occurs in response to a rise in intracellular [Ca++] either after an action potential or in the presence of a hormone/agonist. As depicted in Figure 14-8, a rise in intracellular [Ca++] in smooth muscle results in the binding of 4 Ca++ ions to the protein calmodulin, and then the Ca++-calmodulin complex activates MLCK, which phosphorylates the regulatory light chain of myosin. This phosphorylation step is critical for the
interaction of smooth muscle myosin with actin. In addition to this phosphorylation step in smooth muscle, an ATP molecule is also needed to energize the myosin cross-bridge for the development of force.
|
| page 274 | | | page 275 |
|
| Figure 14-7 Control systems of smooth muscle. Contraction (or inhibition of contraction) of smooth muscles can be initiated by (1) the intrinsic activity of pacemaker cells, (2) neurally released transmitters, or (3) circulating or locally generated hormones or signaling molecules. The combination of a neurotransmitter, hormone, or drug with specific receptors activates contraction by increasing cell Ca++. The response of the cells depends on the concentration of the transmitters or hormones at the cell membrane and the nature of the receptors present. Hormone concentrations depend on diffusion distance, release, reuptake, and catabolism. Consequently, cells lacking close neuromuscular contacts will have a limited response to neural activity unless they are electrically coupled so that depolarization is transmitted from cell to cell. A, Multiunit smooth muscles resemble striated muscles in that there is no electrical coupling and neural regulation is important. B, Single-unit smooth muscles are like cardiac muscle, and electrical activity is propagated throughout the tissue. Most smooth muscles probably lie between the two ends of the single unit-multiunit spectrum. |
|
Table 14-1.
Modulation of Smooth Muscle Activity by Neurotransmitters, Hormones, and Local Factors |
| Agonist | Response | Receptor | Second Messenger |
| Norepinephrine and epinephrine from sympathetic stimulation | Contraction* (predominant)
Relaxation† | α1-AR
β2-AR | InsP3
cAMP |
| Acetylcholine from parasympathetic stimulation | Contraction‡ (direct)
Relaxation‡ (indirect) | Muscarinic receptor on SMC
Muscarinic receptor on EC | |
| Angiotensin II | Contraction§ | AT-II receptor | InsP3 |
| Vasopressin | Contraction§ | Vasopressin receptor | InsP3 |
| Endothelin | Contraction§ | Endothelin receptor | InsP3 |
| Adenosine | Relaxation[Verbar] | Adenosine receptor | cAMP |
|
*The predominant effect of sympathetic stimulation is smooth muscle contraction caused by the abundance of α1-AR relative to β2-AR in smooth muscle.
†Activation of β2-AR on smooth muscle modulates the degree of smooth muscle contraction during sympathetic stimulation. Therapeutic β2-AR agonists are important for the relaxation of bronchial smooth muscle during asthmatic attacks.
‡Vascular smooth muscles are poorly innervated by the parasympathetic system. During vagal stimulation, however, acetylcholine (ACh) can become elevated in the coronary circulation and result in coronary relaxation (mediated by binding of ACh to endothelial cells). Note that this effect of ACh is indirect because binding of ACh to endothelial cells results in release of the smooth muscle relaxant nitric oxide from the endothelial cells. In regions of the coronary circulation with damaged endothelium, binding of ACh to coronary smooth muscle could promote contraction (vasospasm; direct effect).
§A variety of hormones can elevate InsP3 in smooth muscle and thereby result in smooth muscle contraction. Such hormones include angiotensin II, vasopressin, and endothelin, along with the neurotransmitters norepinephrine and acetylcholine. As noted above, however, each hormone/transmitter binds to a specific receptor type.
[Verbar]During periods of intense muscular activity, adenosine can be released from the working muscle, diffuse to the neighboring vasculature, and promote vasodilation. Thus, adenosine is acting as a local factor to increase blood flow to a specific region (i.e., working muscle).
AR, adrenergic receptor; EC, endothelial cell; InsP3, inositol 1,4,5-trisphosphate; SMC, smooth muscle cell.
|
| Contraction of smooth muscle is thus said to be "thick filament regulated," which contrasts with the "thin filament regulation" of contraction of striated muscle, where binding of Ca++ to troponin exposes myosin binding sites on the actin thin filament. The thick filament regulation is attributable to the expression of a distinct myosin isoform in smooth muscle.
|
| page 275 | | | page 276 |
| Figure 14-8 Regulation of smooth muscle myosin interactions with actin by Ca++-stimulated phosphorylation. In the relaxed state, cross-bridges are present as a high-energy myosin-ADP-Pi complex in the presence of ATP. Attachment to actin depends on phosphorylation of the cross-bridge by a Ca++-calmodulin-dependent myosin light-chain kinase (MLCK). Phosphorylated cross-bridges cycle until they are dephosphorylated by myosin phosphatase. Note that cross-bridge phosphorylation at a specific site on a myosin regulatory light chain requires ATP in addition to that used in each cyclic interaction with actin. |
| The myosin cross-bridge cycle in smooth muscle is similar to that in striated muscle in that after attachment to the actin filament, the cross-bridge undergoes a ratchet action in which the thin filament is pulled toward the center of the thick filament and force is generated. ADP and Pi are released from the myosin head at this time, thereby allowing ATP to bind. ATP
decreases the affinity of myosin for actin, which allows the release of myosin from actin. Energy from the newly bound ATP is then used to produce a conformational change in the myosin head (i.e., recocking the head) so that the cross-bridge is ready for another contraction cycle. The cross-bridge cycle continues as long as the myosin cross-bridge remains phosphorylated. Note that although the basic steps of the cross-bridge cycle appear to be the same for striated and smooth muscle, the kinetics of cross-bridge cycling is much slower for smooth muscle.
|
| Cross-bridge cycling continues, with the hydrolysis of 1 ATP molecule per cycle, until myoplasmic [Ca++] falls. With the decrease in [Ca++], MLCK becomes inactive, and the cross-bridges are dephosphorylated by MP (Fig. 14-8).
|
| As indicated in Figure 14-4, the thin filaments in smooth muscle are attached to dense bodies, and the myosin thick filaments appear to reside between two dense bodies and overlap a portion of the thin filaments, much like the overlap of thick and thin filaments in the sarcomere of striated muscle. A bipolar arrangement of myosin molecules within the thick filament is thought to allow the myosin cross-bridges to pull the actin filaments toward the center of the thick filament, thus contracting the smooth muscle and hence developing force.
|
| From a structural standpoint, smooth muscle myosin is similar to striated muscle myosin in that they both contain a pair of heavy chains and two pairs of light chains. Despite this similarity, they represent different gene products and thus have different amino acid sequences. As noted, smooth muscle myosin, unlike skeletal muscle myosin, is unable to interact with the actin thin filament unless the regulatory light chain of myosin is phosphorylated. Moreover, the thin filament in smooth muscle lacks troponin, which plays a critical role in the thin filament regulation of contraction in striated muscle (see Chapter 12).
|
| Although intracellular Ca++ is required for smooth muscle contraction, the sensitivity of contraction to Ca++ is variable. Several hormones/agonists, for example, increase the force of contraction at a given submaximal intracellular [Ca++], thereby resulting in "Ca++ sensitization." A basic mechanism contributing to Ca++ sensitization involves inhibition of MP and a resultant net increase in total myosin light-chain phosphorylation (and hence force) at a given submaximal [Ca++].
|
| Inhibition of MP underlies the phenomenon of Ca++ sensitization that occurs in response to activation of the monomeric G protein RhoA signaling cascade (Fig. 14-9). RhoA activates Rho kinase (ROK), which in turn inhibits MP by both direct and indirect mechanisms. Direct inhibition of MP by activated ROK involves ROK phosphorylation of the myosin-binding subunit (MBS) of MP. Indirect inhibition of MP by activated ROK involves phosphorylation of CPI-17, an endogenous 17-kDa protein, which then inhibits MP. Hormones/agonists such as catecholamines (acting on α1-adrenergic receptors), vasopressin, endothelin, angiotensin, and muscarinic agonists increase the sensitivity of smooth muscle contraction to Ca++ through activation of RhoA/ROK signaling. ROK can also be activated by arachidonic acid and inhibited by Y-27632, a highly specific inhibitor (Fig. 14-9). Though not shown in Figure 14-9, inactive RhoA is typically located in the cytosol, bound to GDP and an inhibitory protein (Rho-GDP dissociation inhibitor [GDI]). Binding of agonist to various G-coupled receptors can activate RhoA by stimulating guanine nucleotide exchange factor (GEF) to yield RhoA-GTP, which localizes to the sarcolemma and activates ROK. |
|
| page 276 | | | page 277 |
| Hyperactivity of the RhoA/ROK signaling cascade has been implicated in various pathological conditions such as hypertension and vasospasm. Hyperactivity of RhoA/ROK in the vascular smooth muscle of hypertensive animals, for example, was manifested by increased
levels of activated RhoA, up-regulation of ROK, enhancement of agonist-induced Ca++ sensitization of contraction, and a greater reduction in blood pressure by ROK inhibitors as compared with normotensive controls. A similar trend was observed in humans in that ROK inhibitors decreased forearm vascular resistance in hypertensive patients to a greater extent than in normotensive controls. ROK inhibitors have also been shown to reverse or prevent experimentally induced cerebral vasospasm and coronary vasospasm, as well as the associated up-regulation of RhoA/ROK and increased myosin light-chain phosphorylation. Hyperactivity of RhoA/ROK has additionally been implicated in bronchial asthma, erectile dysfunction, and preterm labor, as evidenced by the effects of ROK inhibitors. In addition, ROK inhibitors have decreased vascular smooth muscle proliferation and reduced restenosis after balloon angioplasty in rat carotid artery. |
|
| Phasic Versus Tonic Contraction
|
| During a phasic contraction, myoplasmic [Ca++], cross-bridge phosphorylation, and force reach a peak and then return to baseline (Fig. 14-10). In contrast, during a tonic contraction, myoplasmic [Ca++] and cross-bridge phosphorylation decline after an initial spike but do not return to baseline levels. During this later phase, force slowly increases and is sustained at a high level (Fig. 14-10). This sustained force is maintained with only 20% to 30% of the cross-bridges phosphorylated, and thus ATP utilization is reduced. The term "latch state" refers to this condition of tonic contraction during which force is maintained at low energy expenditure.
|
| The latch state is thought to reflect dephosphorylation of the myosin light chain (Fig. 14-11). When the myosin light chain is phosphorylated, the cross-bridges recycle as long as myoplasmic [Ca++] is elevated. However, if an attached cross-bridge is dephosphorylated by MP, the rate of cross-bridge recycling is decreased because detachment of cross-bridges is slower and the myosin light chain must be rephosphorylated before another cycle can begin. When myoplasmic [Ca++] is high, most of the cross-bridges will be phosphorylated (i.e., the MLCK-to-MP activity ratio is high), and shortening velocities or rates of force development will be relatively high. When myoplasmic [Ca++] falls during tonic contractions, the likelihood that a cross-bridge will be dephosphorylated and spend more time in an attached, force-generating conformation increases. However, a low rate of Ca++-dependent phosphorylation of myosin light chains is essential for contraction. The muscle will relax if [Ca++] falls below that required for binding to calmodulin and activation of MLCK (about 0.1 μM).
|
| Energetics and Metabolism
|
| Inappropriate contraction of smooth muscle is associated with many pathological situations. One example is sustained vasospasm of a cerebral artery that develops several hours after a subarachnoid hemorrhage. It is thought that free radicals generated as a result of the hemorrhage raise myoplasmic [Ca++] in surrounding arterial smooth muscle cells. The rise in myoplasmic [Ca++] activates MLCK, which leads to cross-bridge phosphorylation and contraction. The vasoconstriction deprives other areas of the brain of oxygen and may lead to permanent injury or death of surrounding neurons. For a few days the cerebral artery remains sensitive to vasoactive agents, and therefore treatment with vasodilators may restore flow. An increase in ROK activity and MP phosphorylation has been observed during cerebral vasospasm. Administration of ROK inhibitors promotes relaxation of the vasospasm and decreases the level of myosin light-chain phosphorylation. The smooth muscle cells cease to respond to the vasodilators after several days, and they lose contractile proteins and secrete extracellular collagen. The lumen of the artery remains constricted as a result of structural and mechanical changes that do not involve active contraction. |
|
| As already noted, ATP consumption is reduced during the latch state. Under this condition, smooth muscle
uses 300-fold less ATP than would be required by skeletal muscle to generate the same force. Smooth muscle, like skeletal muscle, requires ATP for ion transport to maintain the resting membrane potential, sequester Ca++ in the SR, and extrude Ca++ from the cell. All these metabolic needs are readily met by oxidative phosphorylation. Fatigue of smooth muscle does not occur unless the cell is deprived of oxygen. However, aerobic glycolysis with lactic acid production normally supports membrane ion pumps even when oxygen is plentiful.
|
| REGULATION OF MYOPLASMIC CALCIUM CONCENTRATION
|
| The mechanisms that couple activation to contraction in smooth muscle involve two Ca++ sources: one involving the sarcolemma and the other involving the SR. The sarcolemma regulates Ca++ influx and efflux from the extracellular Ca++ pool. The SR membranes determine Ca++ movement between the myoplasm and the SR pool. Skeletal muscle contraction does not require extracellular Ca++ (see Chapter 12). In contrast, extracellular Ca++ is important for smooth muscle contraction. Thus, regulation of myoplasmic [Ca++] involves not only the SR but also the sarcolemma (Fig. 14-12). A number of factors can alter the myoplasmic [Ca++] of smooth muscle. This differs from skeletal muscle, in which action potential-induced release of Ca++ from the SR fully activates the contractile apparatus.
|
| page 277 | | | page 278 |

|
| Figure 14-9 RhoA/ROK signaling in smooth muscle. A variety of agonists of G-coupled receptors simultaneously stimulate InsP3 production and activate RhoA/ROK signaling. InsP3 is produced by phospholipase C (PLC)-mediated hydrolysis of PIP2. InsP3 increases intracellular [Ca++] by opening InsP3-gated Ca++ channels in the SR, thereby resulting in Ca++-calmodulin-dependent activation of myosin light-chain kinase (MLCK) and subsequent phosphorylation of the myosin regulatory light chain and promotion of actin-myosin interaction (contraction). Activated RhoA (depicted as Rho-GTP) stimulates Rho kinase (ROK), which inhibits myosin phosphatase (MP) by phosphorylating the myosin-binding subunit (MBS) of MP. ROK also inhibits MP indirectly by phosphorylating/activating CPI-17, a 17-kDa inhibitor of MP. The net effect of ROK phosphorylation is a decrease in MP activity, which results in an increased level of myosin light-chain phosphorylation and hence greater force of contraction at a given intracellular [Ca++] (i.e., increased sensitivity of contraction to Ca++). |

|
| Figure 14-10 Time course of events in cross-bridge activation and contraction in smooth muscle. A, A brief period of stimulation is associated with Ca++ mobilization, followed by cross-bridge phosphorylation and cycling to produce a brief phasic, twitch-like contraction. B, In a sustained tonic contraction produced by prolonged stimulation, the Ca++ and phosphorylation levels typically fall from an initial peak. Force is maintained during tonic contractions at a reduced [Ca++] (and hence a low level of myosin light-chain phosphorylation), with lower cross-bridge cycling rates manifested by lower shortening velocities and ATP consumption. |
| page 278 | | | page 279 |
| Figure 14-11 Covalent regulation allows eight cross-bridge states in smooth muscle. Phosphorylation by MLCK (vertical red arrows) is obligatory for cross-bridge attachment. Phosphorylated cross-bridges cycle comparatively rapidly. Dephosphorylation of a cross-bridge during a cycle by a constitutively active MP (vertical black arrows) slows cycling rates and produces the latch state. Calcium regulates cross-bridge cycling by determining phosphorylation rates. Note that ATP is required for both regulation (vertical arrows) and cycling (curved arrows). |

|
| Figure 14-12 Principal mechanisms determining myoplasmic [Ca++] in smooth muscle. Release of calcium from the SR is a rapid initial event in activation, whereas both the SR and the sarcolemma participate in the subsequent stimulus-dependent regulation of myoplasmic [Ca++]. The sarcolemma integrates many simultaneous excitatory and inhibitory inputs to govern the cellular response. Higher-order regulatory mechanisms can alter the activity of various pumps, exchangers, or enzymes (the asterisks designate well-established instances). ATP, process requires ATP hydrolysis; CM, calmodulin; G, guanine nucleotide-binding proteins; IP3, inositol 1,4,5-trisphosphate; MLCK, myosin light-chain kinase; PIP2, phosphatidylinositol bisphosphate; PLC, phospholipase C. |
| page 279 | | | page 280 |
| The role of the smooth muscle SR in regulating myoplasmic [Ca++] is comparable to that of skeletal muscle. Stimulation of the cell opens SR Ca++ channels, and
myoplasmic [Ca++] increases rapidly. This release is not linked to voltage sensors, as is the case in skeletal muscle, but to binding of the second messenger InsP3 to receptors in the SR. InsP3 is generated by a stimulus that acts on sarcolemmal receptors that are coupled via a guanine nucleotide-binding protein (G protein) to activate phospholipase C (PLC) (see Chapter 3). PLC hydrolyzes the membrane phospholipid phosphatidylinositol bisphosphate (PIP2) into InsP3 and diacylglycerol. InsP3 then diffuses to the SR and opens the InsP3-gated Ca++ channel, thereby resulting in release of Ca++ from the SR into the myoplasm. This complex process may permit graded release of Ca++ from the SR and also enable many different neurotransmitters and hormones to effect smooth muscle contraction. Calcium is reaccumulated by the SR through the activity of the SERCA, although as indicated later, extrusion of Ca++ from the smooth muscle cell also contributes to the reduction in myoplasmic [Ca++]. Refilling of the SR with Ca++ not only involves reaccumulation of cytosolic Ca++ but also depends on the extracellular [Ca++]. The dependence on extracellular [Ca++] is thought to reflect the operation of a "store-operated" Ca++ channel present in the sarcolemma at points near underlying SR called "junctional SR."
|
| A variety of hormones and neurotransmitters elevate myoplasmic [Ca++] by stimulating InsP3 production. Vascular smooth muscle, for example, is innervated by sympathetic fibers of the autonomic nervous system. These fibers use norepinephrine as a neurotransmitter, which when released binds to α1-adrenergic receptors on vascular smooth muscle cells and results in G protein-dependent activation of PLC. Activation of PLC results in the production of InsP3, which activates the InsP3-gated Ca++ channel in the SR, thereby elevating myoplasmic [Ca++] and causing vasoconstriction. Other agents that promote vasoconstriction by activating the InsP3 cascade include angiotensin II and vasopressin. The development of drugs that block the production of angiotensin II (e.g., angiotensin-converting enzyme [ACE] inhibitors) provides a means of promoting vasodilation that is important for individuals with hypertension or congestive heart failure. As mentioned previously, a variety of agents can produce contraction of smooth muscle without altering membrane potential (i.e., pharmacomechanical coupling). Agonist-induced activation of the InsP3 cascade represents an example of pharmacomechanical coupling. Many of the hormones/agonists that activate PLC through G protein-coupled receptors also promote sarcolemmal Ca++ influx and activation of RhoA/ROK. The net effect is a rise in intracellular [Ca++], which activates MLCK, concomitant with a rise in ROK activity, which inhibits MP, both of which act complementarily to promote net myosin light-chain phosphorylation.
|
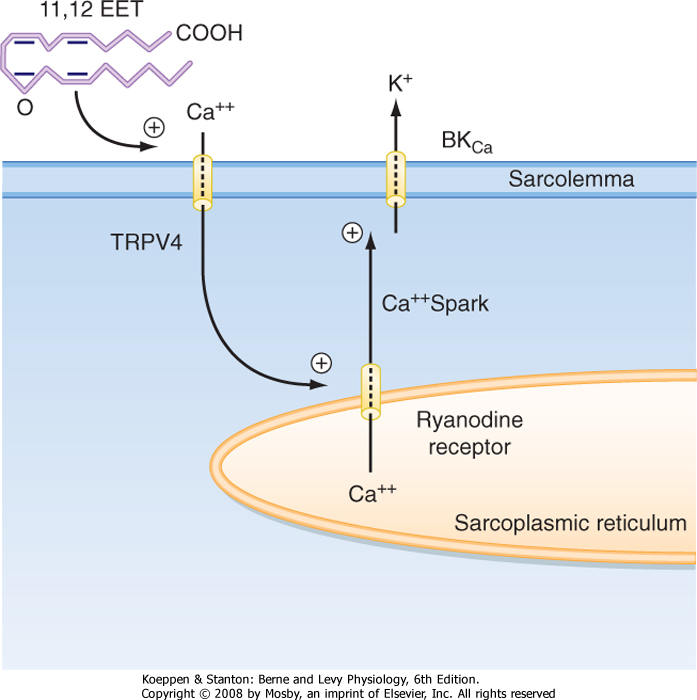
| Calcium sparks have also been observed to occur in smooth muscle in the presence of an endothelial-dependent hyperpolarization factor (EDHF) (Fig. 14-13). Specifically, EDHF appears to be an arachidonic acid metabolite (e.g., epoxyeicosatrienoic acid [EET]) that is produced by endothelial cells in response to various stimuli and then released to the underlying vascular smooth muscle. EET has been shown to activate a transient receptor channel (e.g., TRPV4) in the sarcolemma of smooth muscle that leads to the influx of Ca++, which then opens RYR channels in the SR and results in Ca++ sparks. The Ca++ sparks in turn activate a large-conductance K+ channel in the sarcolemma (BKCa), and the smooth muscle cell becomes hyperpolarized. Hyperpolarization in turn decreases basal Ca++ influx through voltage-gated Ca++ channels in the smooth muscle, thereby decreasing intracellular [Ca++] and hence relaxing the smooth muscle, as described earlier. |
|
|
| Figure 14-13 An arachidonic acid metabolite (11,12-epoxyeicosatrienoic acid [11,12 EET]) released from endothelial cells can open the transient receptor channel TRPV4 in the underlying smooth muscle to permit the influx of Ca++, which in turn initiates brief openings of the SR ryanodine receptor (Ca++ sparks) localized near the sarcolemma. Opening of Ca++-activated K+ channels in the sarcolemma by calcium sparks results in hyperpolarization of the smooth muscle and hence vasodilation. |
| page 280 | | | page 281 |
| In addition to the InsP3 receptor, the SR also contains the Ca++-gated Ca++ channel, also called the RYR, which may be activated during periods of Ca++ influx through the sarcolemma. Short-lived, spontaneous opening of the RYR resulting in localized elevations in myoplasmic [Ca++] occurs in many cells, including
smooth muscle. When observed with Ca++-sensitive fluorescent dyes, these spontaneous localized elevations in myoplasmic [Ca++] produce brief light flashes and as a result are named "Ca++ sparks." In smooth muscle, an increase in cAMP has been associated with
an increase in the frequency of Ca++ sparks, particularly in situations in which the SR is in close proximity to the sarcolemma (i.e., junctional SR, perhaps near caveolae). An increase in the frequency of these sparks hyperpolarizes vascular smooth muscle by activation of a large-conductance Ca++-gated K+ channel in the sarcolemma. This hyperpolarization then decreases overall myoplasmic [Ca++], and relaxation occurs.
|
| Calcium is extruded from the smooth muscle cell by the activity of sarcolemmal Ca++-ATPase and by a 3Na+-1Ca++ antiporter (i.e., 3 Na+ ions enter the cell for each Ca++ ion extruded). Extrusion of Ca++ from the cell competes with sequestration of Ca++ in the SR by SERCA and thus reduces the accumulation of Ca++ in the SR. It is thought that a decrease in SR [Ca++] results in the release of a calcium influx factor (CIF) from the SR, which then activates a "store-operated" Ca++ channel in the sarcolemma near the junctional SR and allows the SR to completely refill with Ca++ from the extracellular fluid. The identity of this CIF and the identity of the store-operated Ca++ channel are not yet known. Nevertheless, it is clear that sustained contraction of smooth muscle requires extracellular Ca++. It has been proposed that Ca++ refilling may occur in the confined space between the caveolae and peripheral SR of smooth muscle.
|
| In addition to the stimulatory effects of various agents on sarcolemma Ca++ channels and InsP3 cascades, there are several inhibitory factors that lower myoplasmic [Ca++] and thereby relax smooth muscle. For example, the dihydropyridine class of Ca++ channel blocking drugs decreases the influx of Ca++ through sarcolemmal L-type voltage-gated Ca++ channels and reduces vasomotor tone. Similarly, drugs that open K+ channels in the sarcolemma (e.g., hydralazine) promote relaxation (e.g., vasodilation) by hyperpolarizing the membrane potential, which reduces the influx of Ca++ through voltage-gated Ca++ channels. Conversely, agents that decrease K+ permeability of the sarcolemma may promote vasoconstriction by inducing membrane depolarization, which then increases influx of Ca++ through these same voltage-gated Ca++ channels. Smooth muscle also contains receptor-activated Ca++ channels. Conductance of these receptor-activated Ca++ channels is linked to receptor occupancy.
|
| A variety of drugs and hormones relax smooth muscle by increasing the cellular concentrations of cAMP or cGMP. Nitric oxide (NO) is produced by nerves and vascular endothelial cells, and it relaxes smooth muscle by increasing cGMP. Acetylcholine released from parasympathetic fibers causes vasodilation in some vascular beds as a result of stimulating the production of NO by vascular endothelial cells. The molecular mechanism or mechanisms underlying the cGMP-dependent relaxation of vascular smooth muscle are complex and may involve activation of a myosin light-chain phosphatase, as well as a reduction in intracellular [Ca++], through stimulation of Ca++ pumps in the sarcolemma or SR, or both. Similarly, elevation of cAMP in vascular smooth muscle by stimulation of β-adrenergic receptors or activation of adenosine receptors promotes vasodilation through cAMP-dependent phosphorylation. In particular, cAMP-dependent phosphorylation of MLCK has been proposed to attenuate the Ca++-dependent increase in MLCK activity, thereby reducing the ability of MLCK to phosphorylate the regulatory light chain of myosin, although cAMP-dependent relaxation also appears to involve a reduction in intracellular [Ca++]. cAMP, for example, has been shown to increase the frequency of Ca++ sparks in smooth muscle, which as described earlier, hyperpolarizes the membrane potential by activation of Ca++-gated K+ channels, thereby reducing the influx of Ca++ through voltage-gated Ca++ channels. Relaxation of smooth muscle by elevation of cAMP has afforded asthmatics a means of reversing bronchiolar constriction through the use of β2-adrenergic agonists. The local vasodilatory effect of adenosine produced in working muscle during periods of intense exercise has also been attributed, at least in part, to elevated cAMP levels in vascular smooth muscle secondary to adenosine-induced stimulation of purinergic receptors on the sarcolemma of vascular smooth muscle. Adenosine may also activate a sarcolemmal K+ channel to induce membrane hyperpolarization, which as already noted will decrease the influx of Ca++ through voltage-gated Ca++ channels and cause vasodilation. Thus, regulation of smooth muscle tone may be under the influence of not only the autonomic nervous system and circulating hormones but also neighboring endothelial cells and skeletal muscle cells via diffusible substances such as NO and adenosine.
|
| DEVELOPMENT AND HYPERTROPHY
|
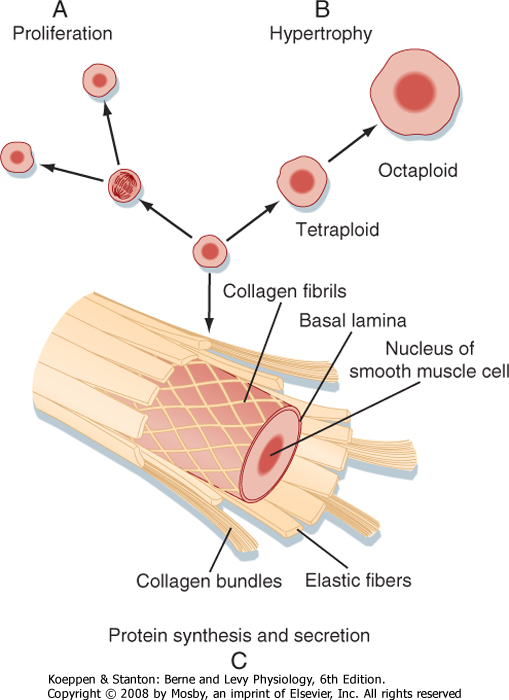
| During development and growth, the number of smooth muscle cells increases (Fig. 14-14). Smooth muscle tissue mass also increases if an organ is subjected to a sustained increase in mechanical work. This increase in mass is called compensatory hypertrophy. A striking example occurs with arterial smooth muscle cells (i.e., in the tunica media of the artery) in hypertensive patients. The increased mechanical load on the muscle cells appears to be the common factor that induces this hypertrophy. Chromosomal replication can result in significant numbers of polyploid muscle cells. The polyploid cells contain multiple sets of the normal number of chromosomes. They synthesize more contractile proteins and thus increase the size of the cell (Fig. 14-14).
|
| page 281 | | | page 282 |
| Figure 14-14 Smooth muscle cells carry out many activities. A, They retain the capacity to divide during normal growth or in certain pathological responses such as the formation of atherosclerotic plaque. B, Cells may also hypertrophy in response to increased loads. Chromosomal replication, not followed by cell division, yields cells with a greater content of contractile proteins. C, Smooth muscle cells also synthesize and secrete the constituents of the extracellular matrix. |
| Although smooth muscle is involved in physiological adjustments to exercise, sustained changes in the mechanical loading that induce cellular adaptations are usually the result of a pathological condition (e.g., hypertension). A fairly common example in men is urinary bladder hypertrophy caused by benign or cancerous enlargement of the prostate gland, which obstructs the bladder outlet. The clinical result is difficulty urinating, distention of the bladder, and impaired emptying. In this situation, the ability of the bladder smooth muscle to contract and develop stress is diminished. The reasons for this remain unexplained, but phenotypic modulation of the smooth muscle cells with altered contractile protein isoform expression and gross anatomic distortion of the bladder wall occurs. Neuromuscular changes also affect myoplasmic Ca++ mobilization and cross-bridge phosphorylation. Fortunately, normal structure and function are usually restored after the obstruction is alleviated. |
|
| The myometrium, which is the smooth muscle component of the uterus, undergoes hypertrophy as parturition (birth) approaches. Hormones play an important role in this response. The smooth muscle is quiescent during pregnancy when the hormone progesterone predominates, and few gap junctions that electrically couple the smooth muscle cells are present. At term, under the dominant influence of estrogen, the myometrium undergoes marked
hypertrophy. Large numbers of gap junctions form just before birth and convert the myometrium to a single-unit tissue to coordinate contraction during parturition.
|
| SYNTHETIC AND SECRETORY FUNCTIONS
|
| The growth and development of tissues that contain smooth muscle are associated with increases in the connective tissue matrix. Smooth muscle cells can synthesize and secrete the materials that make up this matrix, including collagen, elastin, and proteoglycans (Fig. 14-14). The synthetic and secretory capacities are evident when smooth muscle cells are isolated and placed in tissue culture. The cells rapidly lose thick myosin filaments and much of the thin filament lattice, and there is expansion of the rough endoplasmic reticulum and Golgi apparatus. The phenotypically altered cells multiply and lay down connective tissue. This process is reversible, and some degree of redifferentiation with the formation of thick filaments occurs after cell replication ceases. Determinants of the smooth muscle cell phenotype are largely unknown, but hormones and growth factors in blood, as well as mechanical loads on cells, have been implicated in the control of phenotypic modulation.
|
| Atherosclerosis is a disease characterized by lesions located in the wall of blood vessels. The lesions are induced by disorders that injure the endothelium, such as hypertension, diabetes, and smoking. Three formed elements (monocytes, T lymphocytes, and platelets) that circulate in the bloodstream act on the damaged vascular endothelium. There, they generate chemotactic factors and mitogens that modify the structure of the surrounding smooth muscle cells. The latter lose most of their thick and thin filaments and develop an extensive rough endoplasmic reticulum and Golgi complex. These cells migrate to the subendothelial space (i.e., the tunica media of the artery), proliferate, and participate in formation of the fatty lesions or the fibrous plaques that characterize atherosclerosis. Inhibition or down-regulation of Rho kinase (ROK) has been shown to promote the regression of atherosclerotic-like lesions in an animal model. The mechanism or mechanisms underlying this beneficial effect of ROK inhibition are unclear but may be related to the regulation of both endothelial permeability and monocyte migration by ROK. That is, hyperactivity of ROK has been implicated in various pathological conditions, including increased transendothelial permeability (perhaps secondary to increased actomyosin activity), whereas inhibition of ROK has been shown to decrease transendothelial migration of monocytes and neutrophils. |
|
| page 282 | | | page 283 |
| BIOPHYSICAL PROPERTIES OF SMOOTH MUSCLE
|
| Length-Tension Relationship
|
| Smooth muscle contains large amounts of connective tissue composed of extensible elastin fibrils and inextensible collagen fibrils. Because this extracellular matrix can withstand high distending forces or loads, it is responsible for the passive length-tension curve measured in relaxed tissues. This ability of the matrix also limits organ volume.
|
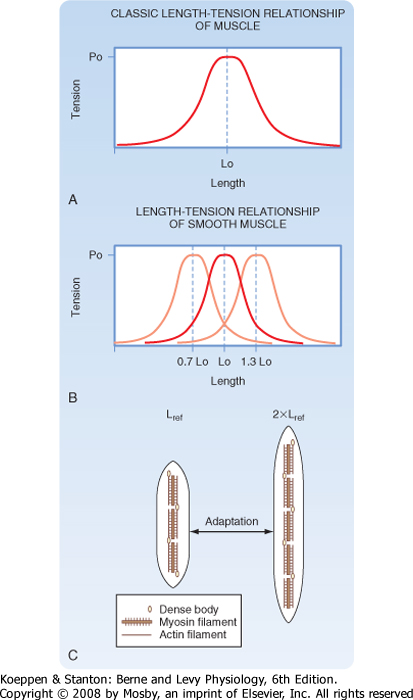
| When lengths are normalized to the optimal length for the development of force (i.e., L0), the length-tension curves for smooth and skeletal muscle are very similar (Fig. 14-15; see also Chapter 12). However, the length-tension curves of striated and smooth muscle differ quantitatively. For example, smooth muscle cells shorten more than skeletal muscle cells do. In addition, smooth muscle is characteristically only partially activated, and the peak isometric force attained varies with the stimulus. In skeletal muscle, the stimulus (i.e., action potential) always produces a full twitch contraction. Smooth muscle can generate active force comparable to that of skeletal muscle, even though smooth muscle contains only about a fourth as much myosin. This does not imply that the cross-bridges in smooth muscle have greater force-generating capacity. Instead, active cross-bridges in smooth muscle are much more likely to be in the attached, force-generating configuration because of their slow cycling kinetics.
|
| Smooth muscle has the unique ability to shift the length-tension curve, depending on the resting length. Thus, if the smooth muscle is stretched, the length-tension curve will shift to longer lengths over the course of tens of minutes to hours (see Fig. 14-15, B). Similarly, if the smooth muscle is allowed to return to a shorter resting length, the length-tension relationship will shift to the left, again over a period of tens of minutes to hours, depending on the stimulation frequency. This unusual property of smooth muscle is termed "length adaptation." The molecular basis for this change in the length-tension relationship depending on the resting length of the muscle is thought to involve an alteration in the number of contractile units in series (see Fig. 14-15, C).
|
| Force-Velocity Relationship
|
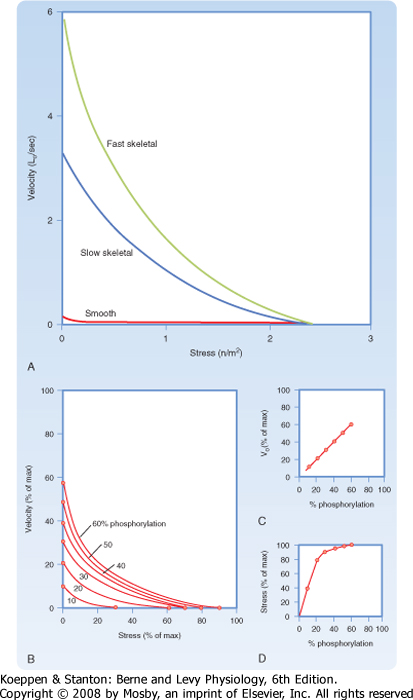
| Smooth and striated muscles both exhibit a hyperbolic dependence of shortening velocity on load. However, contraction velocities are far slower in smooth muscle than in striated muscle. One factor that underlies these slow velocities is that the myosin isoform in smooth muscle cells has low ATPase activity.
|
| Figure 14-15 Length adaptation of smooth muscle. Both skeletal muscle (A) and smooth muscle (B) exhibit a bell-shaped length-tension relationship, although the length-tension relationship of smooth muscle can vary. Within a short period after stretching smooth muscle, there is a rightward shift in the length-tension relationship such that maximal force generation occurs at a longer muscle length (B). Likewise, within a brief time after shortening of a smooth muscle, there is a leftward shift in the length-tension relationship (B). The mechanism or mechanisms underlying this length adaptation are hypothesized to reflect a change in the number of contractile units in series (C). |
| page 283 | | | page 284 |
| Figure 14-16 A, Force-velocity curves for fast and slow human skeletal muscle cells and smooth muscle. B, Smooth muscles have variable force-velocity relationships that are determined by the level of Ca++-stimulated cross-bridge phosphorylation. C, Maximal shortening velocities with no load (intercepts on the ordinate in B) are directly dependent on cross-bridge phosphorylation by MLCK. D, Active force/stress (abscissa intercepts in B) rises rapidly with phosphorylation and, near maximal stress, may be generated with only 20% to 30% of the cross-bridges in the phosphorylated state. |
| page 284 | | | page 285 |
| Skeletal muscle cells have a force-velocity curve in which shortening velocities are determined only by load and the myosin isoform (see Chapter 12). In contrast, both force and shortening velocity, which reflect the number of cycling cross-bridges and their cycling rates, vary in smooth muscle. When activation of smooth muscle is altered, for example, by different
frequencies of nerve stimulation or changing hormone concentrations, a "family" of velocity-stress curves can be derived (Fig. 14-16). This implies that both cross-bridge cycling rates and the number of active cross-bridges in smooth muscle are regulated in some
way, which is in marked contrast to striated muscle. This difference is conferred by a regulatory system that depends on the phosphorylation of cross-bridges, which in turn depends on myoplasmic [Ca++]. Because myosin light-chain phosphorylation is required for actin-myosin interaction in smooth muscle, a dependence of maximal force on the degree of myosin phosphorylation is expected (i.e., phosphorylation of more myosin molecules results in more actin-myosin interactions and hence more force generated). The variation in maximal shortening velocity as a function of the degree of myosin phosphorylation may reflect
dephosphorylation of the myosin light chain while the myosin is still attached to the actin, thus slowing the rate of detachment (i.e., latch state) at low levels of phosphorylation. At higher levels of phosphorylation, the likelihood of latch states would be reduced and the myosin cross-bridges would be released more quickly from actin, thereby yielding a higher shortening velocity at all loads (see Fig. 14-16B).
|
- Smooth muscle cells are linked by a variety of junctions that serve both mechanical and communication roles. These linkages are essential in cells that must contract uniformly.
- The sarcolemma plays an important role in Ca++ exchange between the extracellular fluid and the myoplasm. The sarcolemma of smooth muscle contains numerous caveolae that contribute to the regulation of intracellular [Ca++] and also appear to serve as a scaffold for signaling molecules. The SR contains an intracellular Ca++ pool that can be mobilized to transiently increase myoplasmic [Ca++]. Myoplasmic [Ca++] is dependent on extracellular Ca++. Transporters in the sarcolemma that regulate myoplasmic [Ca++] include receptor-mediated Ca++ channels, voltage-gated Ca++ channels, Ca++-ATPase, and the 3Na+-1Ca++ antiporter. The SR also regulates myoplasmic [Ca++]. The Ca++ channels in the SR open in response to a chemical. Neurotransmitters or hormones that act via receptors in the sarcolemma can activate PLC, followed by generation of the second messenger InsP3. InsP3 then activates InsP3-gated Ca++ channels on the SR. Many agonists that activate PLC through G protein-coupled receptors also activate the RhoA/ROK signaling cascade, thereby increasing the sensitivity of smooth muscle contraction to Ca++. Smooth muscle SR also contains Ca++-gated Ca++ channels (RYR). Ca++ reaccumulates in the SR via SERCA.
- Smooth muscles contain contractile units that consist of small groups of thick myosin filaments that interdigitate with large numbers of thin filaments attached to Z line equivalents termed dense bodies or membrane-dense areas. No striations are evident. Contraction is caused by a sliding filament-cross-bridge mechanism.
- Contraction of smooth muscle is dependent on both release of Ca++ from the SR and entry of Ca++ across the sarcolemma. Smooth muscle lacks troponin. Phosphorylation of cross-bridges by a Ca++-dependent MLCK is necessary for attachment to the thin filament. Dephosphorylation of an attached cross-bridge by MP slows its cycling rates. Higher myoplasmic [Ca++] increases the ratio of MLCK to MP activity, with the result that more of the cross-bridges remain phosphorylated throughout a cycle. This increases shortening velocities.
- Smooth muscle activity is controlled by nerves (principally autonomic), circulating hormones, locally generated signaling substances, junctions with other smooth muscle cells, and even junctions with other non-smooth muscle cells. A variety of hormones/agonists increase the sensitivity of smooth muscle contraction to Ca++ by reducing the activity of MP. Activation of the RhoA/ROK signaling cascade contributes to this inhibition of MP and hence to the increase in sensitivity of smooth muscle contraction to Ca++.
- The response to sustained or tonic stimulation is a rapid contraction followed by sustained maintenance of force with reduced cross-bridge cycling rates and ATP consumption. This behavior, called the latch state, is advantageous for muscles that may need to withstand continuous external force, such as blood vessels, which must be able to withstand blood pressure. During the latch state, ATP is consumed at less than 1/300 the rate needed to maintain the same force in skeletal muscle.
- The length-tension relationships, hyperbolic velocity-load relationships, power output curves, and ability to resist imposed loads are comparable to those of skeletal muscle. Shortening velocities and ATP consumption rates are very low in smooth muscle, in keeping with expression of a myosin isoform with low activity. Uniquely, smooth muscle has the ability to adjust the length-tension relationship when chronically stretched or shortened, a process termed "length adaptation." Smooth muscles also have the unusual ability to alter velocity-stress relationships, which reflects regulation of both the number of active cross-bridges (determining force) and their average cycling rates for a given load (determining velocity).
- Smooth muscle is also a synthetic and secretory cell with a major role in formation of the extensive extracellular matrix that surrounds and links the cells. Cellular hypertrophy occurs in response to physiological needs, and smooth muscle cells retain the potential to divide.
|
|
| page 285 | | | page 286 |
|