| 18 Regulation of the Heart and Vasculature
|
| REGULATION OF HEART RATE AND CONTRACTILITY
|
Cardiac output is defined as the quantity of blood pumped by the heart each minute. Cardiac output may be varied by changing the heart rate or the volume of blood ejected from either ventricle with each heartbeat; this volume is called the stroke volume. Mathematically, cardiac output (CO) can be expressed as the product of heart rate (HR) and stroke volume (SV):

|
| Thus, understanding how cardiac activity is controlled can be gained by considering how the heart rate and stroke volume are regulated. Heart rate is regulated by the activity of the cardiac pacemaker, and stroke volume is directly related to myocardial performance. These two determinants are interdependent because a change in one determinant of cardiac output almost invariably alters the other determinant.
|
| NERVOUS CONTROL OF THE HEART RATE
|
| Although certain local factors, such as temperature changes and stretching of tissue, can affect the heart rate, the autonomic nervous system is the principal means by which the heart rate is controlled.
|
| The average resting heart rate is about 70 beats/min in normal adults, and it is significantly greater in children. During sleep the heart rate decelerates by 10 to 20 beats/min, and during emotional excitement or muscular activity it may accelerate to rates well above 100. In well-trained athletes the resting rate is usually only about 50 beats/min.
|
| Both divisions of the autonomic nervous system tonically influence the cardiac pacemaker, which is normally the sinoatrial (SA) node. The sympathetic system enhances automaticity, whereas the parasympathetic system inhibits it. Changes in heart rate usually involve a reciprocal action of these two divisions of the autonomic nervous system. Thus, the heart rate ordinarily increases with a combined decrease in parasympathetic activity and increase in sympathetic activity; the heart rate decreases with the opposite changes in autonomic neural activity.
|
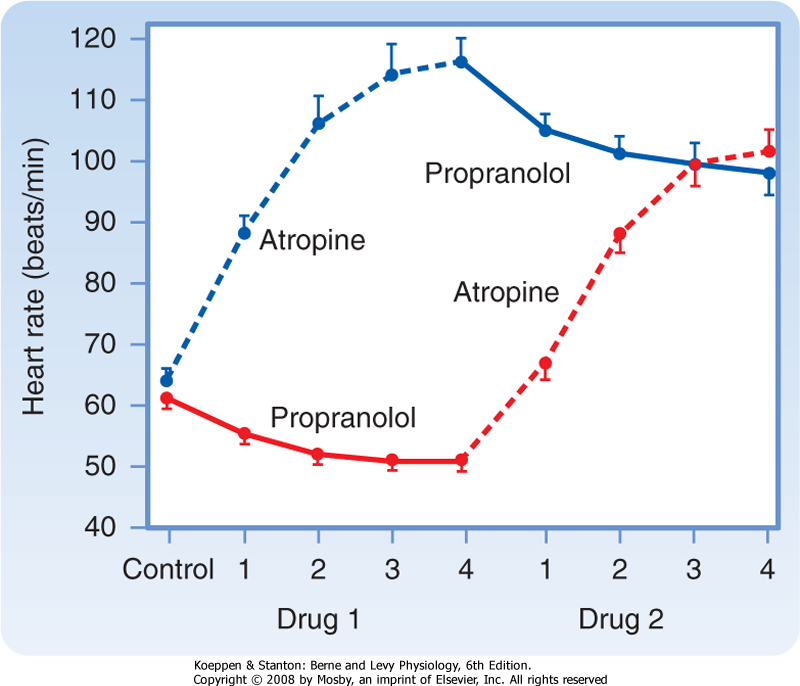
| Parasympathetic tone usually predominates in healthy, resting individuals. When a resting individual is given atropine, a muscarinic receptor antagonist that blocks parasympathetic effects, the heart rate generally increases substantially. If a resting individual is given propranolol, a β-adrenergic receptor antagonist that blocks sympathetic effects, the heart rate usually decreases only slightly (Fig. 18-1). When both divisions of the autonomic nervous system are blocked, the heart rate of young adults averages about 100 beats/min. The rate that prevails after complete autonomic blockade is called the intrinsic heart rate.
|
| The cardiac parasympathetic fibers originate in the medulla oblongata, in cells that lie in the dorsal motor nucleus of the vagus or in the nucleus ambiguus (see Chapter 11). The precise location of the parasympathetic fibers varies among species. In humans, centrifugal vagal fibers pass inferiorly through the neck near the common carotid arteries and then through the mediastinum to synapse with postganglionic vagal cells. These cells are located either on the epicardial surface or within the walls of the heart. Most of the vagal ganglion cells are located in epicardial fat pads near the SA and atrioventicular (AV) nodes.
|
| The right and left vagi are distributed to different cardiac structures. The right vagus nerve affects the SA node predominantly; stimulation of this nerve slows SA nodal firing and can even stop the firing for several seconds. The left vagus nerve mainly inhibits AV conduction tissue to produce various degrees of AV block (see Chapter 16). However, the distribution of the efferent vagal fibers is overlapping such that left vagal stimulation also depresses the SA node and right vagal stimulation impedes AV conduction.
|
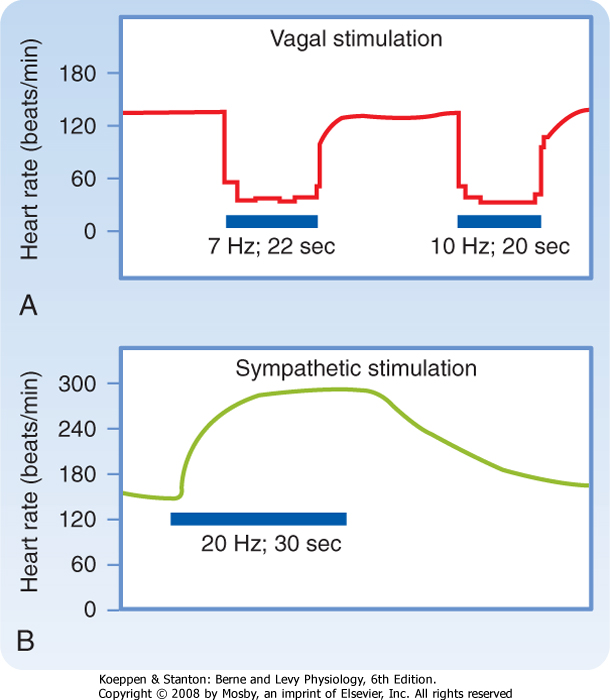
| The SA and AV nodes are rich in cholinesterase, an enzyme that rapidly hydrolyzes the neurotransmitter acetylcholine (ACh). The effects of a given vagal stimulus decay very quickly (Fig. 18-2, A) when vagal stimulation is discontinued because ACh is rapidly destroyed. In addition, vagal effects on SA and AV nodal function have a very short latency (≈50 to 100 msec) because the ACh released quickly activates special ACh-regulated K+ channels (KACh) in the cardiac cells. These channels open quickly because the muscarinic receptor is coupled directly to the KACh channel by a guanine nucleotide-binding protein (Gi). These two features of the vagus nerves-brief latency and rapid decay of the response-permit them to exert beat-by-beat control of SA and AV nodal function.
|
| page 370 |  | | page 371 |
| Figure 18-1 Effects of four equal doses of atropine (muscarinic receptor antagonist that blocks parasympathetic effects) and propranolol (β-adrenergic receptor antagonist that blocks sympathetic effects) on the heart rate of 10 healthy young men. In half of the trials, atropine was given first (top curve); in the other half, propranolol was given first (bottom curve). (Redrawn from Katona PG et al: J Appl Physiol 52:1652, 1982.) |
| Figure 18-2 Changes in heart rate evoked by stimulation (horizontal bars) of the vagus (A) and sympathetic nerves (B). (Modified from Warner HR, Cox A: J Appl Physiol 17:349, 1962.) |
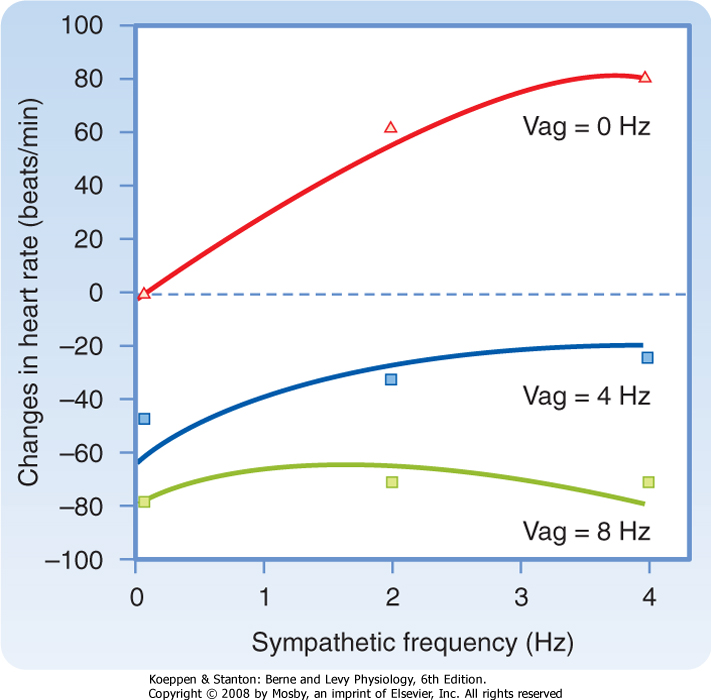
| Figure 18-3 Changes in heart rate when the vagus and cardiac sympathetic nerves are stimulated simultaneously. The sympathetic nerves are stimulated at 0, 2, and 4 Hz and the vagus nerves at 0, 4, and 8 Hz. (Modified from Levy MN, Zieske H: J Appl Physiol 27:465, 1969.) |
| Parasympathetic influences usually predominate over sympathetic effects at the SA node, as shown in Figure 18-3. When the frequency of sympathetic stimulation increases from 0 to 4 Hz, the heart rate increases by about 80 beats/min in the absence of vagal stimulation (Vag = 0 Hz). However, when the vagi are stimulated at 8 Hz, increasing the sympathetic stimulation
frequency from 0 to 4 Hz has only a negligible influence on heart rate.
|
| The cardiac sympathetic fibers originate in the intermediolateral columns of the upper five or six thoracic and lower one or two cervical segments of the spinal cord (see Chapter 11). These fibers emerge from the spinal column through the white communicating branches and enter the paravertebral chains of ganglia. The preganglionic and postganglionic neurons synapse mainly in the stellate or middle cervical ganglia, depending on the species. In the mediastinum, the postganglionic and preganglionic parasympathetic fibers join to form a complicated plexus of mixed efferent nerves to the heart.
|
| The postganglionic cardiac sympathetic fibers in this plexus approach the base of the heart along the adventitial surface of the great vessels. From the base of the heart, these fibers are distributed to the various chambers as an extensive epicardial plexus. They then penetrate the myocardium, usually accompanying the coronary vessels.
|
| page 371 | | | page 372 |
| In contrast to abrupt termination of the response after vagal activity, the effects of sympathetic stimulation decay gradually after stimulation is stopped (Fig. 18-2, B). Nerve terminals take up to 70% of the norepinephrine released during sympathetic stimulation; much of the remainder is carried away by the bloodstream. These processes are slow. Furthermore, the facilitatory effects of sympathetic stimulation on the heart attain steady-state values much more slowly than do the inhibitory effects of vagal stimulation. The onset of the cardiac response to sympathetic stimulation begins slowly for two main reasons. First, norepinephrine appears to be released slowly from the
sympathetic nerve terminals. Second, the cardiac effects of the neurally released norepinephrine are mediated mainly by a relatively slow second messenger system involving cAMP (see Chapter 3). Hence, sympathetic activity alters the heart rate and AV conduction much more slowly than vagal activity does. Although vagal activity can exert beat-by-beat control of cardiac function, sympathetic activity cannot.
|
| Control by Higher Centers
|
| Stimulation of various brain regions can have significant effects on cardiac rate, rhythm, and contractility (see Chapter 11). In the cerebral cortex, centers that regulate cardiac function are located in the anterior half of the brain, principally in the frontal lobe, the orbital cortex, the motor and premotor cortex, the anterior portion of the temporal lobe, the insula, and the cingulate gyrus. Stimulation of the midline, ventral, and medial nuclei of the thalamus elicits tachycardia. Stimulation of the posterior and posterolateral regions of the hypothalamus can also change the heart rate. Stimuli applied to the H2 fields of Forel in the diencephalon evoke various cardiovascular responses, including tachycardia; these changes resemble those observed during muscular exercise. Undoubtedly, the cortical and diencephalic centers initiate the cardiac reactions that occur during excitement, anxiety, and other emotional states. The hypothalamic centers also initiate the cardiac response to alterations in environmental temperature. Experimentally induced temperature changes in the preoptic anterior hypothalamus alter the heart rate and peripheral resistance.
|
| Stimulation of the parahypoglossal area of the medulla reciprocally activates cardiac sympathetic pathways and inhibits cardiac parasympathetic pathways. In certain dorsal regions of the medulla, distinct cardiac accelerator (increase the heart rate) and augmentor (increase cardiac contractility) sites have been detected in animals with transected vagi. The accelerator regions are more abundant on the right side, whereas the augmentor sites are more prevalent on the left. A similar distribution also exists in the hypothalamus. Therefore, the sympathetic fibers mainly descend ipsilaterally through the brainstem.
|
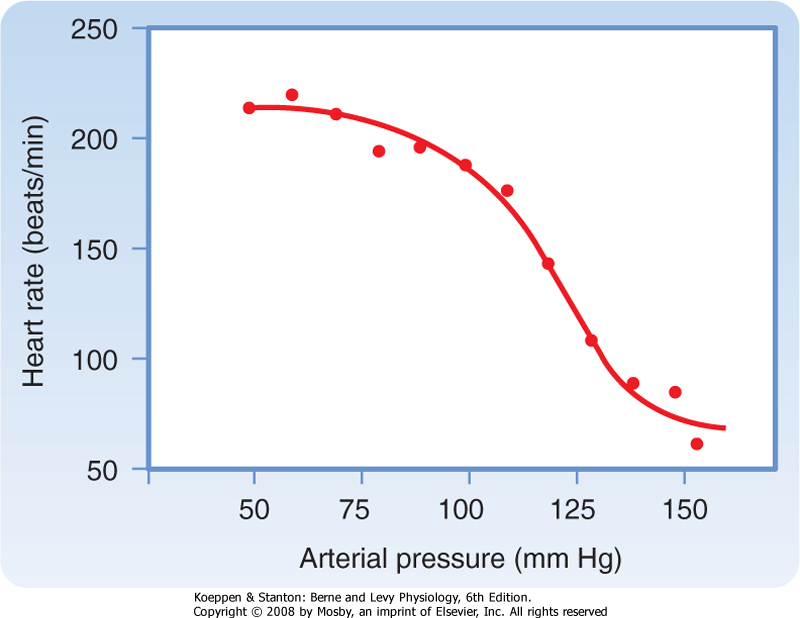
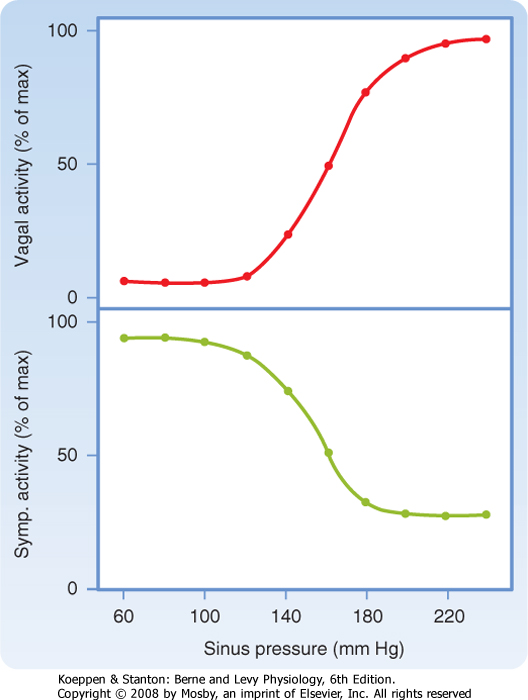
| Sudden changes in arterial blood pressure initiate a reflex that evokes an inverse change in heart rate (Fig. 18-4). Baroreceptors located in the aortic arch and carotid sinuses are responsible for this reflex. The inverse relationship between heart rate and arterial blood pressure is generally most pronounced over an intermediate range of arterial blood pressure. Below this intermediate range, the heart rate maintains a constant, high value; above this pressure range, the heart rate maintains a constant, low value.
|
| Figure 18-4 Heart rate as a function of mean arterial pressure. |
| Figure 18-5 Effects of changes in pressure in isolated carotid sinuses on neural activity in cardiac vagal and sympathetic efferent nerve fibers. (Adapted from Kollai M, Koizumi K: Pflügers Arch 413:365, 1989.) |
| The effects of these changes in carotid sinus pressure on activity in the cardiac autonomic nerves are presented in Figure 18-5, which shows that over an intermediate range of carotid sinus pressure (100 to 180 mm Hg), reciprocal changes are evoked in efferent vagal and sympathetic neural activity. Below this range
of carotid sinus pressure, sympathetic activity is intense and vagal activity is virtually absent. Conversely, above the intermediate range of carotid sinus pressure, vagal activity is intense and sympathetic activity is minimal.
|
| Bainbridge Reflex, Atrial Receptors, and Atrial Natriuretic Peptide
|
| page 372 | | | page 373 |
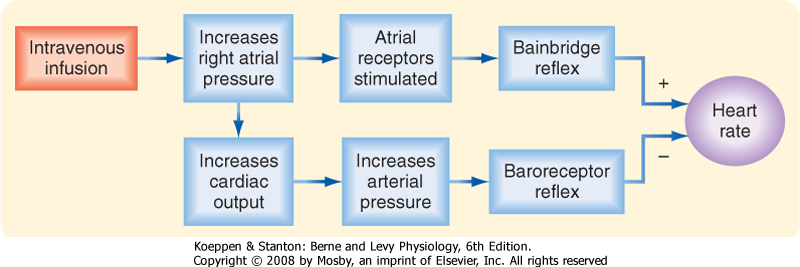
| Figure 18-6 Intravenous infusions of blood or electrolyte solutions tend to increase the heart rate via the Bainbridge reflex and to decrease the heart rate via the baroreceptor reflex. The actual change in heart rate induced by such infusions is the result of these two opposing effects. |
| In 1915, Bainbridge reported that infusing blood or saline into dogs accelerated their heart rate. This
increase did not seem to be tied to arterial blood pressure because the heart rate rose regardless of whether arterial blood pressure did or did not change. However, Bainbridge also noted that the heart rate increased whenever central venous pressure rose sufficiently to distend the right side of the heart. Bilateral transection of the vagi abolished this response. This is termed the Bainbridge reflex.
|
| Many investigators have confirmed Bainbridge's observations and have noted that the magnitude and direction of the response depend on the prevailing heart rate. When the heart rate is slow, intravenous infusions usually accelerate the heart. At more rapid heart rates, however, infusions ordinarily slow the heart. What accounts for these different responses? Increases in blood volume not only evoke the so-called Bainbridge reflex but also activate other reflexes (notably the baroreceptor reflex). These other reflexes tend to elicit opposite changes in heart rate. Therefore, changes in heart rate evoked by an alteration in blood volume are the result of these antagonistic reflex effects (Fig. 18-6). Evidently, the Bainbridge reflex predominates over the baroreceptor reflex when blood volume rises, but the baroreceptor reflex prevails over the Bainbridge reflex when blood volume diminishes.
|
| Both atria have receptors that are affected by changes in blood volume and that influence the heart rate. These receptors are located principally in the venoatrial junctions: in the right atrium at its junctions with the venae cavae and in the left atrium at its junctions with the pulmonary veins. Distention of these atrial receptors sends afferent impulses to the brainstem in the vagi. The efferent impulses are carried from the brainstem to the SA node by fibers from both autonomic divisions.
|
| The cardiac response to these changes in autonomic neural activity is highly selective. Even when the reflex increase in heart rate is large, changes in ventricular contractility are generally negligible. Furthermore, the neurally induced increase in heart rate is not usually accompanied by an increase in sympathetic activity in the peripheral arterioles.
|
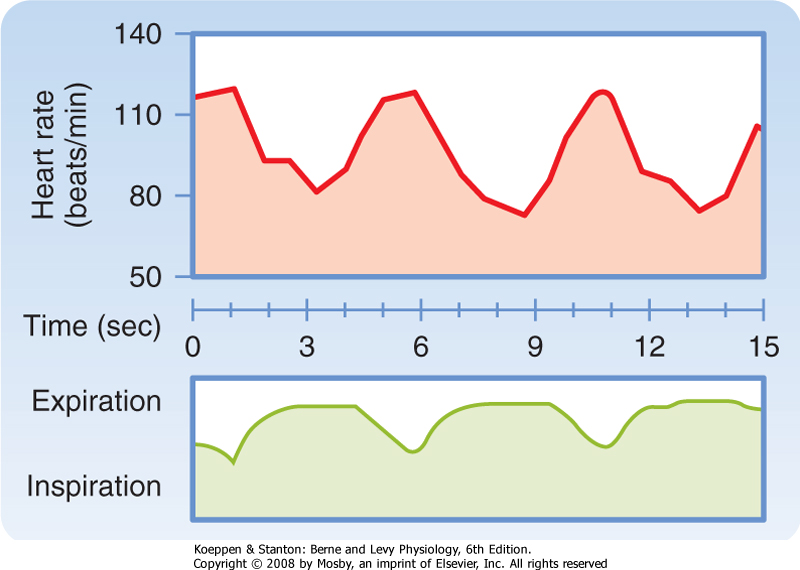
| Figure 18-7 Respiratory sinus arrhythmia. Note that the cardiac cycle length increases during expiration and decreases during inspiration. (Modified from Warner MR et al: Am J Physiol 251:H1134, 1986.) |
| In congestive heart failure, NaCl and water are retained, mainly because stimulation by the renin-angiotensin system increases the release of aldosterone from the adrenal cortex. The plasma level of ANP is also increased in congestive heart failure. By enhancing the renal excretion of NaCl and water, this peptide gradually reduces fluid retention and the consequent elevations in central venous pressure and cardiac preload. |
|
| Stimulation of the atrial receptors increases not only the heart rate but also urine volume. Reduced activity in the renal sympathetic nerve fibers may partially account for this diuresis. However, the principal mechanism appears to be a neurally mediated reduction in vasopressin (antidiuretic hormone) secretion by the posterior pituitary gland (see Chapters 34 and 40). Stretch of the atrial walls also releases atrial natriuretic
peptide (ANP) from the atria.* ANP, a 28-amino acid peptide, exerts potent diuretic and natriuretic effects on the kidneys (see also Chapter 34) and vasodilator effects on the resistance and capacitance vessels. Thus, ANP is an important regulator of blood volume and blood pressure.
|
| Respiratory Sinus Arrhythmia
|
| Rhythmic variations in heart rate, occurring at the frequency of respiration, are detectable in most individuals and tend to be more pronounced in children. The heart rate typically accelerates during inspiration and decelerates during expiration (Fig. 18-7).
|
| page 373 | | | page 374 |
| Recordings from cardiac autonomic nerves reveal that neural activity increases in the sympathetic fibers during inspiration and increases in the vagal fibers during expiration. The heart rate response to cessation of vagal stimulation is very quick because as already noted, the ACh released from the vagus nerves is rapidly hydrolyzed by cholinesterase. This short latency permits the heart rate to vary rhythmically at the respiratory frequency. Conversely, the norepinephrine released periodically at the sympathetic endings is removed very slowly. Therefore, the rhythmic variations in sympathetic activity that accompany inspiration do not induce any appreciable oscillatory changes in heart rate. Thus, respiratory sinus arrhythmia is almost entirely brought about by changes in vagal activity. In fact, respiratory sinus arrhythmia is exaggerated when vagal tone is enhanced.
|
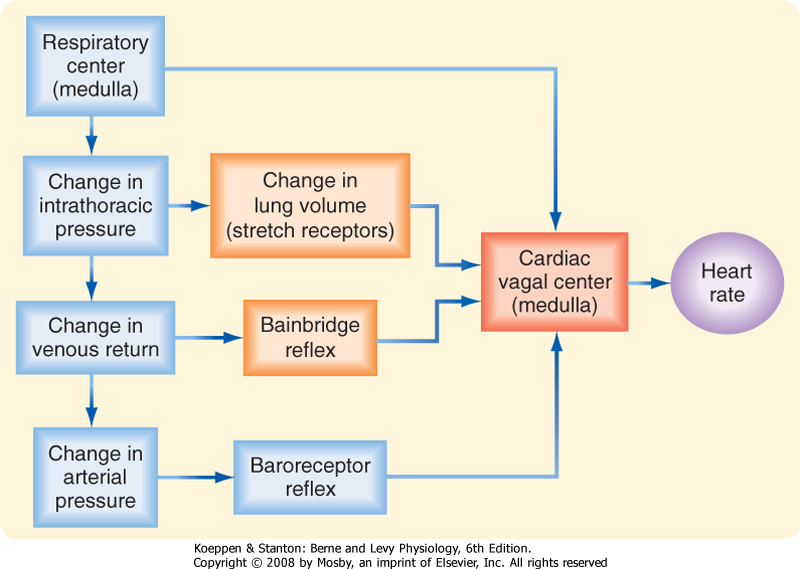
| Both reflex and central factors help initiate respiratory sinus arrhythmia (Fig. 18-8). Stretch receptors in the lungs are stimulated during inspiration, and this action leads to a reflex increase in heart rate. The afferent and efferent limbs of this reflex are located in the vagus nerves. Intrathoracic pressure also decreases during inspiration and thereby increases venous return to the right side of the heart (see Chapter 19). The consequent stretch of the right atrium elicits the Bainbridge reflex. After the time delay required for the increased venous return to reach the left side of the heart, left ventricular output increases and raises arterial blood pressure. This rise in blood pressure in turn reduces the heart rate through the baroreceptor reflex.
|
|
| Figure 18-8 Respiratory sinus arrhythmia is generated by a direct interaction between the respiratory and cardiac centers in the medulla, as well as by reflexes that originate from stretch receptors in the lungs, stretch receptors in the right atrium (the Bainbridge reflex), and baroreceptors in the carotid sinuses and aortic arch. |
| Central factors are also responsible for respiratory cardiac arrhythmia. The respiratory center in the medulla directly influences the cardiac autonomic centers (Fig. 18-8). In heart-lung bypass experiments, the chest is open, the lungs are collapsed, venous return is diverted to a pump-oxygenator, and arterial blood pressure is maintained at a constant level. In such experiments, rhythmic movement of the rib cage
attests to the activity of the medullary respiratory centers. Such movement of the rib cage is often accompanied by rhythmic changes in heart rate at the respiratory frequency. This respiratory cardiac arrhythmia is almost certainly induced by a direct interaction between the respiratory and cardiac centers in the medulla.
|
| The cardiac response to peripheral chemoreceptor stimulation illustrates the complex interactions that may ensue when one stimulus excites two organ systems simultaneously. In intact animals, stimulation of the carotid chemoreceptors consistently increases ventilatory rate and depth (see Chapter 24), but ordinarily it changes the heart rate only slightly. The magnitude of the ventilatory response determines whether the heart rate increases or decreases as a result of carotid chemoreceptor stimulation. Mild chemoreceptor-induced stimulation of respiration decreases the heart rate moderately; more pronounced stimulation increases the heart rate only slightly. If the pulmonary response to chemoreceptor stimulation is blocked, the heart rate response may be greatly exaggerated, as described later.
|
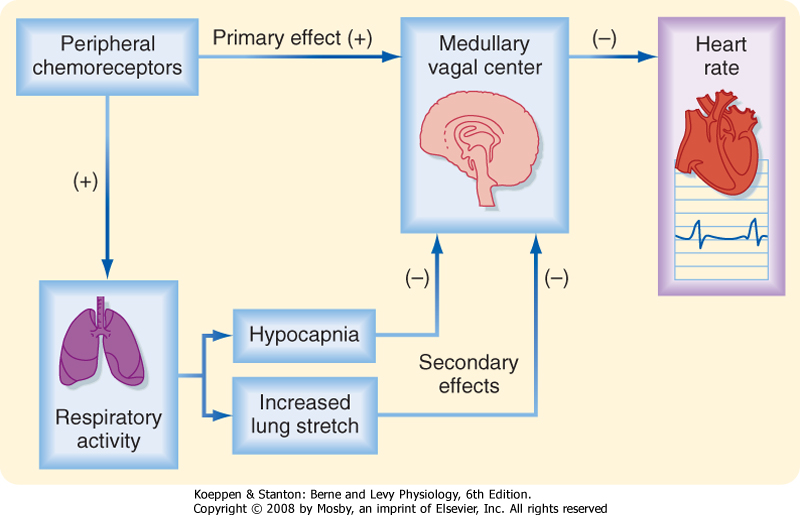
| The cardiac response to peripheral chemoreceptor stimulation is the result of primary and secondary reflex mechanisms (Fig. 18-9). The principal effect of the primary reflex stimulation is to excite the medullary vagal center and thereby decrease the heart rate. The respiratory system mediates secondary reflex effects. The respiratory stimulation by arterial chemoreceptors tends to inhibit the medullary vagal center. This inhibition varies with the level of concomitant stimulation of respiration; small increases in respiration inhibit the vagal center slightly, whereas large increases in ventilation inhibit the vagal center more profoundly.
|
| page 374 | | | page 375 |
| Figure 18-9 The primary effect of stimulation of peripheral chemoreceptors on the heart rate is to excite the cardiac vagal center in the medulla and thus to decrease the heart rate. Peripheral chemoreceptor stimulation also excites the respiratory center in the medulla. This effect produces hypocapnia and increases lung inflation, both of which secondarily inhibit the medullary vagal center. Thus, these secondary influences attenuate the primary reflex effect of peripheral chemoreceptor stimulation on heart rate. |
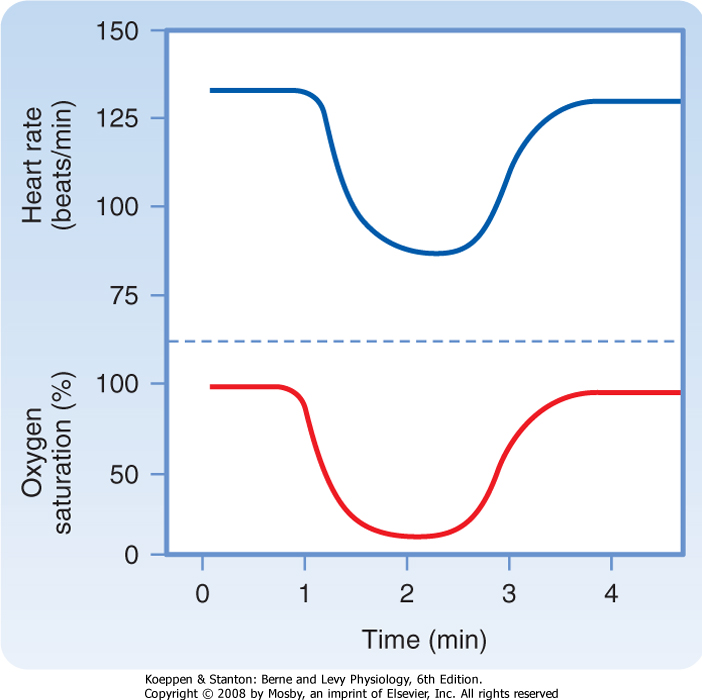
| Figure 18-10 Changes in heart rate with carotid chemoreceptor stimulation during total heart bypass. The lungs remain deflated and respiratory gas exchange is accomplished by an artificial oxygenator. The lower tracing represents the oxygen saturation of the blood perfusing the carotid chemoreceptors. The blood perfusing the remainder of the body, including the myocardium, is fully saturated with oxygen. (Modified from Levy MN et al: Circ Res 18:67, 1966.) |
| An example of the primary inhibitory influence is shown in Figure 18-10. In this example, the lungs are
completely collapsed and blood oxygenation is accomplished with an artificial oxygenator. When the carotid chemoreceptors are stimulated, an intense bradycardia and some degree of AV block ensue. Such effects are mediated primarily by efferent vagal fibers.
|
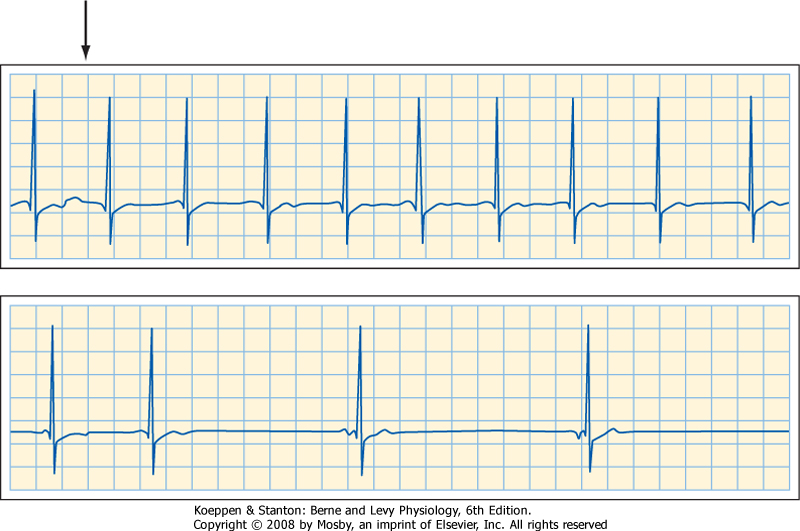
| The electrocardiogram in Figure 18-11 was recorded from a quadriplegic patient who could not breathe spontaneously and required tracheal intubation and artificial respiration. When the tracheal catheter was briefly disconnected (near the beginning of the top strip in the figure) to permit nursing care, profound bradycardia quickly developed. The patient's heart rate was 65 beats/min just before the tracheal catheter was disconnected. In less than 10 seconds after cessation of artificial respiration, his heart rate dropped to about 20 beats/min. This bradycardia could be prevented by blocking the effects of efferent vagal activity with atropine, and its onset could be delayed considerably by hyperventilating the patient before disconnecting the tracheal catheter. |
|
| The pulmonary hyperventilation that is ordinarily evoked by carotid chemoreceptor stimulation influences the heart rate secondarily, both by initiating more pronounced pulmonary inflation reflexes and by producing hypocapnia (Fig. 18-9). Both influences tend to depress the primary cardiac response to chemoreceptor stimulation and thereby accelerate the heart. Hence, when pulmonary hyperventilation is not prevented, the primary and secondary effects neutralize
each other, and carotid chemoreceptor stimulation affects the heart rate only moderately.
|
| Ventricular Receptor Reflexes
|
| Sensory receptors located near the endocardial surfaces of the ventricles initiate reflex effects similar to those elicited by the arterial baroreceptors. Excitation of these endocardial receptors diminishes the heart rate and peripheral resistance. Other sensory receptors have been identified in the epicardial regions of the ventricles. Although all these ventricular receptors are excited by various mechanical and chemical stimuli, their exact physiological functions remain unclear.
|
| REGULATION OF MYOCARDIAL PERFORMANCE
|
| Intrinsic Regulation of Myocardial Performance
|
| page 375 | | | page 376 |
| Figure 18-11 Electrocardiogram of a 30-year-old quadriplegic man who could not breathe spontaneously and required tracheal intubation and artificial respiration. The two strips are continuous. (Modified from Berk JL, Levy MN: Eur Surg Res 9:75, 1977.) |
| Ventricular receptors have been implicated in the initiation of vasovagal syncope, a feeling of lightheadedness or brief loss of consciousness that may be triggered by psychological or orthostatic stress. The ventricular receptors are believed to be stimulated by reduced ventricular filling volume combined with vigorous ventricular contraction. In a person standing quietly, ventricular filling is diminished because blood tends to pool in the veins in the abdomen and legs, as explained in Chapter 17. Consequently, the reduction in cardiac output and arterial blood pressure leads to a generalized increase in sympathetic neural activity via the baroreceptor reflex (Fig. 18-5). The enhanced sympathetic activity to the heart evokes a vigorous ventricular contraction that thereby stimulates the ventricular receptors. Excitation of the ventricular receptors is believed to initiate the autonomic neural changes that evoke vasovagal syncope, namely, a combination of a profound, vagally mediated bradycardia and generalized arteriolar vasodilation mediated by a reduction in sympathetic neural activity. |
|
| The heart is partially or completely denervated in various clinical situations: (1) a surgically transplanted heart is totally denervated, although the intrinsic, postganglionic parasympathetic fibers persist; (2) atropine blocks vagal effects on the heart, and propranolol blocks sympathetic β-adrenergic influences; (3) certain drugs, such as reserpine, deplete cardiac norepinephrine stores and thereby restrict or abolish sympathetic control; and (4) in chronic congestive heart failure, cardiac norepinephrine stores are often severely diminished, and any sympathetic influences are attenuated. |
|
| As noted previously, the heart can initiate its own beat in the absence of any nervous or hormonal control.
The myocardium can also adapt to changing hemodynamic conditions by means of mechanisms that are intrinsic to cardiac muscle itself. For example, racing greyhounds with denervated hearts perform almost as well as those with intact innervation. Their maximal running speed decreases by only 5% after complete cardiac denervation. In these dogs, the threefold to fourfold increase in cardiac output during a race is achieved principally by an increase in stroke volume. Normally, the increase in cardiac output with exercise is accompanied by a proportionate increase in heart rate; stroke volume does not change much (see Chapter 19). This adaptation in the denervated heart is not achieved entirely by intrinsic mechanisms; circulating
catecholamines undoubtedly contribute. For example, if β-adrenergic receptor antagonists are given to greyhounds with denervated hearts, their racing performance is severely impaired.
|
| Two principal intrinsic mechanisms, namely, the Frank-Starling mechanism and rate-induced regulation, enable the myocardium to adapt to changes in hemodynamic conditions. The Frank-Starling mechanism (Starling's law of the heart) is invoked in response to changes in the resting length of myocardial fibers. Rate-induced regulation is evoked by changes in the frequency of the heartbeat.
|
| About a century ago, the German physiologist Otto Frank and the English physiologist Ernest Starling independently studied the response of isolated hearts to changes in preload and afterload (see Chapter 16). When ventricular filling pressure (preload) is increased, ventricular volume increases progressively and after several beats attains a constant, larger volume. At equilibrium, the volume of blood ejected by the ventricles (stroke volume) with each heartbeat increases to equal the greater quantity of venous return to the right atrium.
|
| page 376 | | | page 377 |
| The increased ventricular volume facilitates ventricular contraction and enables the ventricles to pump a greater stroke volume. This increase in ventricular volume is associated with an increase in length of the individual ventricular cardiac fibers. The increase in fiber length alters cardiac performance mainly by altering the number of myofilament cross-bridges that interact (see Chapter 16). More recent evidence indicates that the principal mechanism involves a stretch-induced change in the sensitivity of cardiac myofilaments to Ca++ (see Chapter 16). An optimal fiber length exists, however. Excessively high filling pressures that overstretch the myocardial fibers may depress rather than enhance the pumping capacity of the ventricles.
|
| Starling also showed that isolated heart preparations could adapt to changes in the counterforce to the ventricular ejection of blood during systole (i.e., afterload). As the left ventricle contracts, it does not eject blood into the aorta until the ventricle has developed a pressure that just exceeds the prevailing aortic pressure (see Chapter 16). The aortic pressure during ventricular ejection essentially constitutes the left ventricular afterload. In Starling's experiments, arterial pressure was controlled by a hydraulic device in the tubing that led from the ascending aorta to the right atrial blood reservoir. Venous return to the right atrium was held constant by maintaining the hydrostatic level of the blood reservoir. As Starling raised arterial pressure to a new, constant level, the left ventricle responded at first to the increased afterload by pumping a diminished stroke volume. Because venous return was held constant, the diminution in stroke volume was accompanied by a rise in ventricular diastolic volume, as well as by an increase in the length of the myocardial fibers. This change in end-diastolic fiber length finally enabled the ventricle to pump a normal stroke volume against the greater peripheral resistance. Again, a change in the number of cross-bridges between the thick and thin filaments probably contributes to this adaptation, but the major factor appears to be a stretch-induced change in the sensitivity of the contractile proteins to Ca++.
|
| Cardiac adaptation to alterations in heart rate also involves changes in ventricular volume. During bradycardia, for example, the increased duration of diastole permits greater ventricular filling. The consequent increase in myocardial fiber length increases stroke volume. Therefore, the reduction in heart rate may be fully compensated by the increase in stroke volume, and cardiac output may therefore remain constant.
|
| When cardiac compensation involves ventricular dilation, one must consider how the increased size of the ventricle affects the generation of intraventricular pressure. According to the Laplace relationship (see Chapter 17), if the ventricle enlarges, the force required by each myocardial fiber to generate a given intraventricular systolic pressure must be appreciably greater than that developed by the fibers in a ventricle of normal size. Thus, more energy is required for a dilated heart to perform a given amount of external work than for a normal-sized heart. Hence, computation of afterload on contracting myocardial fibers in the walls of the ventricles must consider ventricular dimensions along with intraventricular (and aortic) pressure.
|
| The relatively rigid pericardium that encloses the heart determines the pressure-volume relationship at high levels of pressure and volume. The pericardium limits heart volume even under normal conditions, when an individual is at rest and the heart rate is slow. In patients with chronic congestive heart failure, the sustained cardiac dilation and hypertrophy may stretch the pericardium considerably. In such patients, the pericardial limitation of cardiac filling is exerted at pressures and volumes entirely different from those in normal individuals.
|
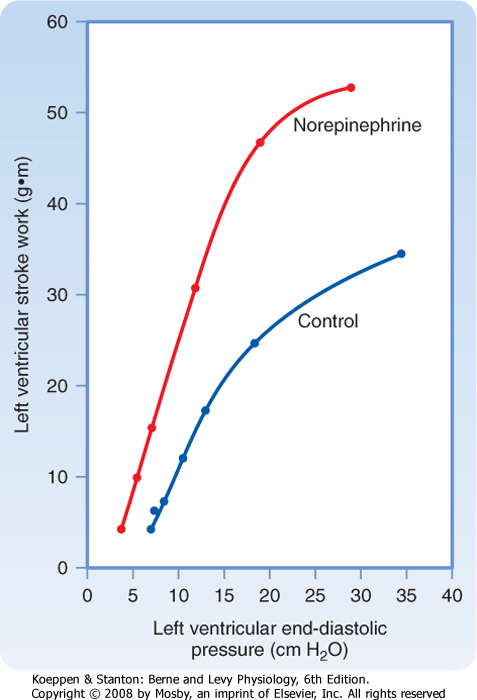
| Figure 18-12 A constant infusion of norepinephrine shifts the ventricular function curve to the left. This shift signifies an enhancement in ventricular contractility. (Redrawn from Sarnoff SJ et al: Circ Res 8:1108, 1960.) |
| page 377 | | | page 378 |
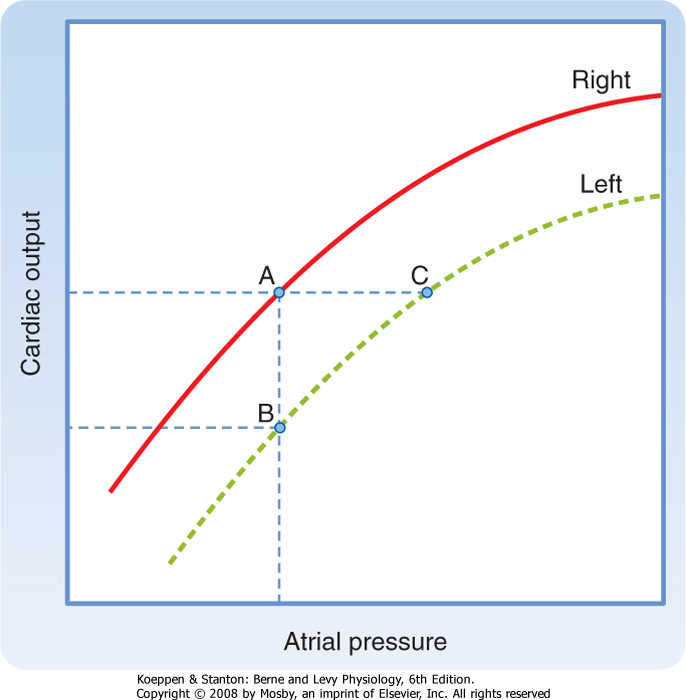
| Figure 18-13 Relationships between the output of the right and left ventricles and mean pressure in the right and left atria, respectively. At any given level of cardiac output, mean left atrial pressure (e.g., point C) exceeds mean right atrial pressure (point A). |
| To assess changes in ventricular performance, the Frank-Starling mechanism is often represented by a family of ventricular function curves. To construct a control ventricular function curve, for example, blood volume is altered over a range of values, and stroke work (i.e., stroke volume × mean arterial pressure) and end-diastolic ventricular pressure are measured at each step. Similar observations are then made during the desired experimental intervention. For example, the ventricular function curve obtained during infusion of norepinephrine lies above and to the left of the control ventricular function curve (Fig. 18-12). Clearly, for a given level of left ventricular end-diastolic pressure (an index of preload), the left ventricle performs more work during the norepinephrine infusion than
during control conditions. Hence, the upward and leftward shift of the ventricular function curve signifies improved ventricular contractility. Conversely, a shift downward and to the right indicates impaired contractility and a tendency toward cardiac failure.
|
| Balance between Right and Left Ventricular Output
|
| The Frank-Starling mechanism is well suited to match cardiac output to venous return. Any sudden, excessive output by one ventricle soon causes an increase in venous return to the second ventricle. The consequent increase in diastolic fiber length in the second ventricle augments the output of that ventricle to correspond to the output of its mate. In this way, the Frank-Starling mechanism maintains a precise balance between the output of the right and left ventricles. Because the two ventricles are arranged in series in a closed circuit, any small, but maintained imbalance in output of the two ventricles would otherwise be catastrophic.
|
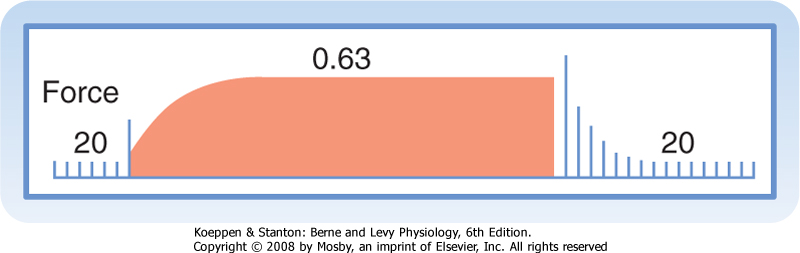
| Figure 18-14 Changes in development of force in an isolated papillary muscle from a cat as the interval between contractions is varied from 20 seconds to 0.63 second and then back to 20 seconds. (Redrawn from Koch-Weser J, Blinks JR: Pharmacol Rev 15:601, 1963.) |
| This greater left than right atrial pressure accounts for the observation that in individuals with congenital atrial septal defects in which the two atria communicate with each other via a patent foramen ovale, the direction of shunt flow is usually from left to right. |
|
| The curves that relate cardiac output to mean atrial pressure for the two ventricles do not coincide; the curve for the left ventricle usually lies below that for the right ventricle (Fig. 18-13). At equal right and left atrial pressure (points A and B), right ventricular output exceeds left ventricular output. Hence, venous return to the left ventricle (a function of right ventricular output) exceeds left ventricular output, and left ventricular diastolic volume and pressure rise. By the Frank-Starling mechanism, left ventricular output therefore increases (from B toward C). Only when the output of both ventricles is identical (points A and C) is equilibrium reached. Under such conditions, however, left atrial pressure (C) exceeds right atrial
pressure (A). This is precisely the relationship that ordinarily prevails.
|
| Myocardial performance is also regulated by changes in the frequency at which the myocardial fibers contract. The effects of changes in contraction frequency on the force developed in an isometrically contracting papillary muscle are shown in Figure 18-14. Initially, the cardiac muscle strip is stimulated to contract once every 20 seconds. When the muscle is suddenly made to contract once every 0.63 second, the force developed increases progressively over the next several beats. At the new steady state, the force developed is more than five times as great as at the larger contraction interval. A return to the larger interval (20 seconds) has the opposite influence on the development of force.
|
| The rise in the force developed when the contraction interval is decreased is caused by a gradual increase in [Ca++]i. Two mechanisms contribute to the rise in [Ca++]: an increase in the number of depolarizations per minute and an increase in the inward Ca++ current per depolarization.
|
| In the first mechanism, Ca++ enters the myocardial cell during each action potential plateau (see Chapter 16). As the interval between beats is diminished, the number of plateaus per minute increases. Although the duration of each action potential (and of each plateau) decreases as the interval between beats is reduced, the overriding effect of the increased number of plateaus per minute on the influx of Ca++ prevails, and [Ca++]i increases.
|
| page 378 | | | page 379 |
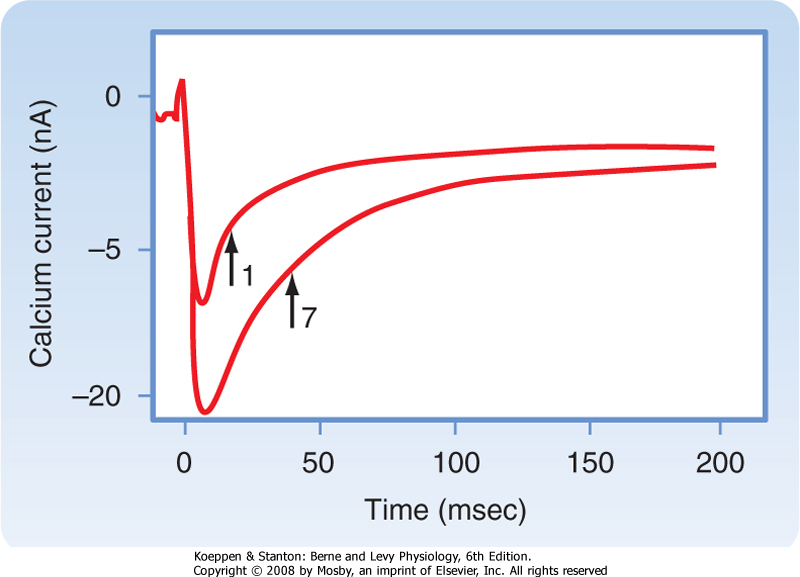
| In the second mechanism, as the interval between beats is suddenly diminished, the inward Ca++ current (iCa) progressively increases with each successive beat until a new steady state is attained at the new basic cycle length. In an isolated ventricular myocyte, influx
of Ca++ into the myocyte increases on successive depolarizations (Fig. 18-15). Both the increased magnitude and the slowed inactivation of iCa result in greater Ca++ influx into the myocyte during the later depolarizations than during the first depolarization. This greater Ca++ influx strengthens contraction.
|
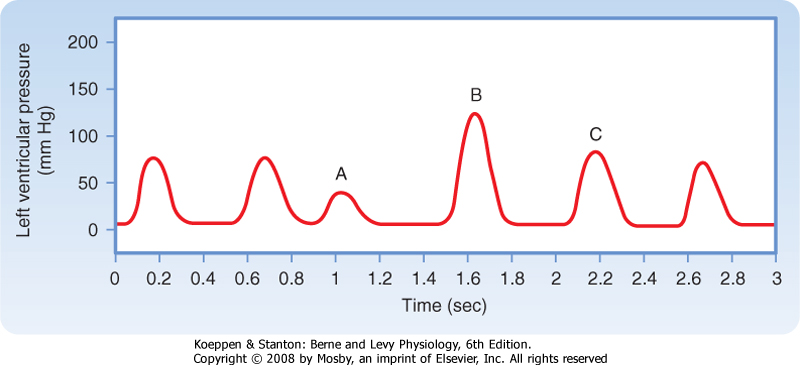
| Transient changes in the intervals between beats also profoundly affect the strength of contraction. When the left ventricle contracts prematurely (Fig. 18-16, beat A), the premature contraction (extrasystole) itself is weak, whereas contraction B (postextrasystolic contraction) after the compensatory pause is very strong. In the intact circulatory system, this response depends partly on the Frank-Starling mechanism. Inadequate time for ventricular filling just before the premature beat results in the weak premature contraction. Subsequently, the exaggerated degree of filling associated with the long compensatory pause (Fig. 18-16, beat B) contributes to the vigorous postextrasystolic contraction.
|
|
| Figure 18-15 Calcium currents induced in a myocyte during the first and seventh depolarizations in a consecutive sequence of depolarizations. The arrows indicate the half-times of inactivation. Note that during the seventh depolarization, the maximal inward Ca++ current and the half-time of inactivation were greater than the respective values for the first depolarization. (Modified from Lee KS: Proc Natl Acad Sci U S A 84:3941, 1987.) |
| Figure 18-16 In an isovolumic left ventricle preparation, a premature ventricular systole (beat A) is typically weak, whereas the postextrasystolic contraction (beat B) is characteristically strong, and the enhanced contractility may diminish over a few beats (e.g., contraction C). (From Levy MN: Unpublished tracing.) |
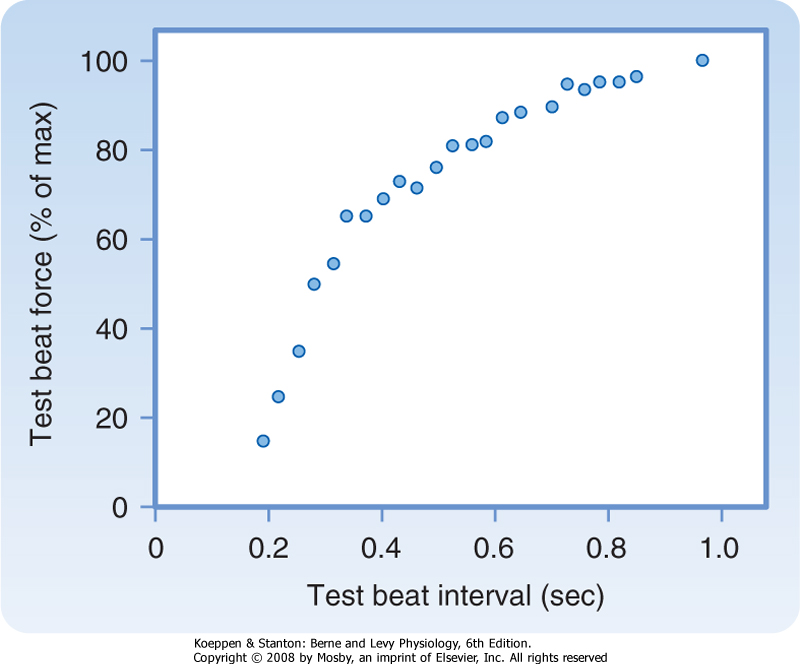
| The weakness of the premature beat is directly related to its degree of prematurity. Thus, the earlier
the premature beat, the weaker its force of contraction. The curve that relates strength of contraction of a premature beat to the coupling interval is called a mechanical restitution curve. Figure 18-17 shows the restitution curve obtained by varying the coupling intervals of test beats in an isolated ventricular muscle preparation.
|
| Figure 18-17 Force generated during premature contractions in an isolated ventricular muscle preparation. The muscle was stimulated to contract once per second. Periodically, the muscle was stimulated prematurely. The scale along the x axis denotes the time between the driven and the premature beat. The y axis denotes the ratio of the contractile force of the premature beat to that of the driven beat. (Modified from Seed WA, Walker JM: Cardiovasc Res 22:303, 1988.) |
| page 379 | | | page 380 |
| Figure 18-18 In an isovolumic left ventricle preparation, stimulation of cardiac sympathetic nerves evokes a substantial rise in peak left ventricular pressure and in the maximal rates of rise and fall in intraventricular pressure (dP/dt). (From Levy MN: Unpublished tracing.) |
| Restitution of the force of contraction depends on the time course of the intracellular circulation of Ca++ in cardiac myocytes during contraction and relaxation. During relaxation, the Ca++ that dissociates from the contractile proteins is taken up by the sarcoplasmic reticulum for subsequent release. However, there is a lag of about 500 to 800 msec before this Ca++ is available for release from the sarcoplasmic reticulum in response to the next depolarization. Thus, the strength of the premature beat is reduced because the time during the preceding relaxation is insufficient to allow much of the Ca++ taken up by the sarcoplasmic reticulum to become available for release during the
premature beat. Conversely, the postextrasystolic beat is considerably stronger than normal because more Ca++ is released from the sarcoplasmic reticulum as a result of the relatively large amount of Ca++ taken up by it during the time that had elapsed from the end of the last regular beat until the beginning of the postextrasystolic beat.
|
| Extrinsic Regulation of Myocardial Performance
|
| Although a completely isolated heart can adapt well to changes in preload and afterload, various extrinsic factors also influence the heart in an individual. Often, these extrinsic regulatory mechanisms may overwhelm the intrinsic mechanisms. The extrinsic regulatory factors may be subdivided into nervous and chemical components.
|
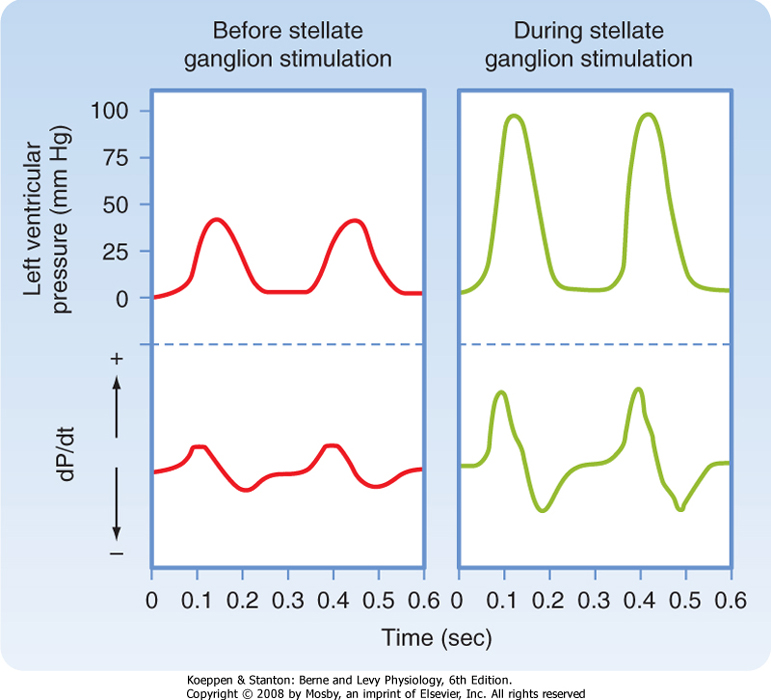
| Sympathetic nervous activity enhances atrial and ventricular contractility. The alterations in ventricular contraction evoked by electrical stimulation of the left stellate ganglion in a isovolumic left ventricle preparation are shown in Figure 18-18. Note that the duration of systole is reduced and the rate of ventricular relaxation is increased during the early phases of diastole; both these effects assist ventricular filling. For any given cardiac cycle length, the abbreviated systole allows more time for diastole and hence for ventricular filling.
|
|
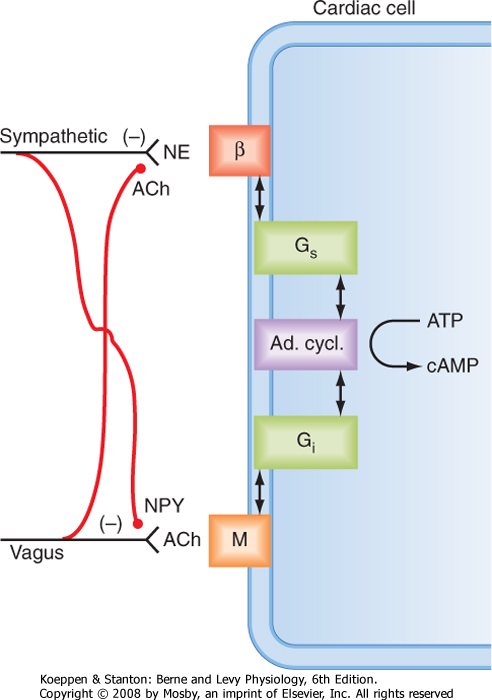
| Figure 18-19 Interneuronal and intracellular mechanisms responsible for interactions between the sympathetic and parasympathetic systems in the neural control of cardiac function. ACh, acetycholine; Ad cycl, adenylyl cyclase; β, β-adrenergic receptor; Gs and Gi, stimulatory and inhibitory G proteins; M, muscarinic receptor; NE, norepinephrine; NPY, neuropeptide Y. (From Levy MN: In Kulbertus HE, Franck G [eds]: Neurocardiology. Mt. Kisco, NY, Futura, 1988.) |
| page 380 | | | page 381 |
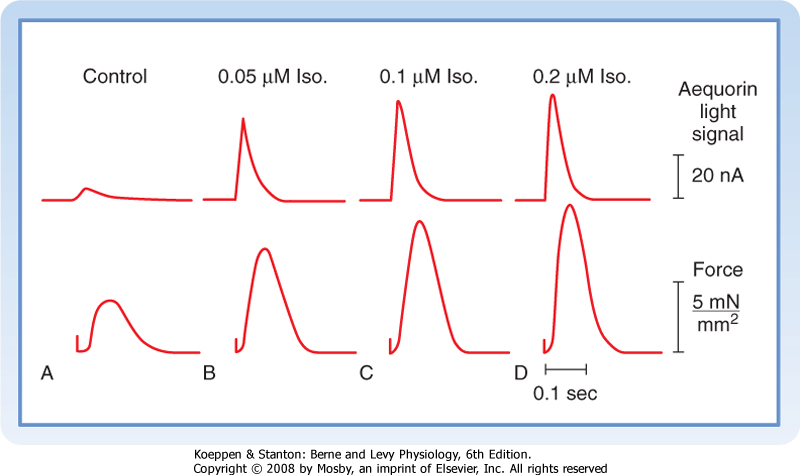
| Figure 18-20 Effects of various concentrations of isoproterenol (Iso) on the aequorin light signal (in nA) and contractile force (in mN/mm2) in a rat ventricular muscle injected with aequorin. The aequorin light signal reflects the instantaneous changes in intracellular [Ca++]. (Modified from Kurihara S, Konishi M: Pflügers Arch 409:427, 1987.) |
| Sympathetic nervous activity also enhances myocardial performance by altering intracellular Ca++ dynamics (see Chapter 16). Neurally released norepinephrine or circulating catecholamines interact with β-adrenergic receptors on the cardiac cell membranes (Fig. 18-19). This interaction activates adenylyl cyclase, which raises intracellular levels of cAMP (see Chapter 3). Consequently, protein kinases that promote the phosphorylation of various proteins are activated
within the myocardial cells. Phosphorylation of phospholamban facilitates reuptake of Ca++ by the sarcoplasmic reticulum, and phosphorylation of troponin I reduces the sensitivity of contractile proteins to Ca++. These effects facilitate relaxation and reduce end-diastolic
pressure (see Chapter 19). Phosphorylation of specific sarcolemmal proteins also activates Ca++ channels in the membranes of myocardial cells.
|
| Activation of Ca++ channels increases the influx of Ca++ during the action potential plateau, and more Ca++ is released from the sarcoplasmic reticulum in response to each cardiac excitation. The contractile strength of the heart is thereby increased. Figure 18-20 shows the correlation between the contractile force in a thin strip of ventricular muscle and the free [Ca++] (indicated by the aequorin light signal) in the myoplasm as the concentration of isoproterenol (a β-adrenergic agonist) is increased.
|
| The overall effect of increased cardiac sympathetic activity in intact animals can best be appreciated in terms of families of ventricular function curves. When the frequency of electrical stimulation applied to the left stellate ganglion increases, the ventricular function curves shift progressively to the left. The changes parallel those produced by infusions of norepinephrine (Fig. 18-12). Hence, for any given left ventricular end-diastolic pressure, the ventricle can perform more work as sympathetic nervous activity is increased.
|
| Parasympathetic Influences
|
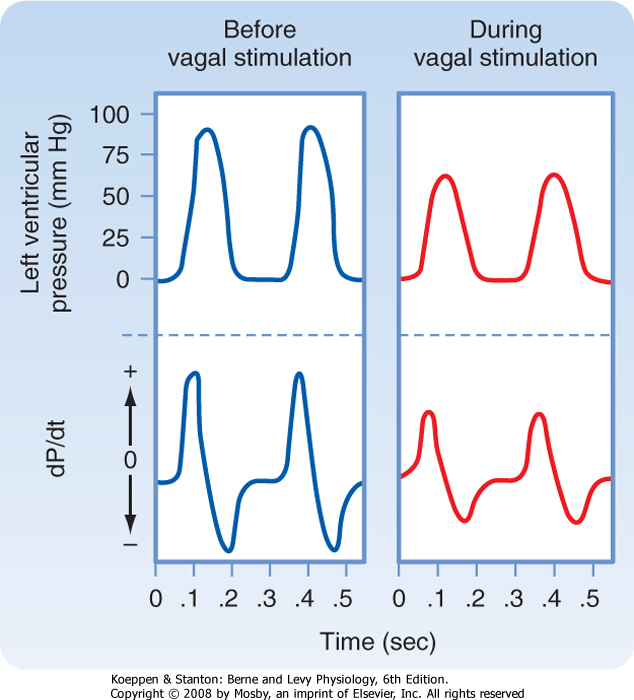
| The vagus nerves inhibit the cardiac pacemaker, atrial myocardium, and AV conduction tissue. The vagus nerves also depress the ventricular myocardium, but the effects are less pronounced than in the atria. In isovolumic left ventricle preparations, vagal stimulation decreases peak left ventricular pressure, the maximal rate of pressure development (dP/dt), and the maximal rate of pressure decline during diastole (Fig. 18-21). In pumping heart preparations, the ventricular function curve shifts to the right during vagal stimulation.
|
| Figure 18-21 In an isovolumic left ventricle preparation, when the ventricle is paced at a constant frequency, vagal stimulation decreases peak left ventricular pressure and diminishes the maximal rates of rise and fall in pressure (dP/dt). (From Levy MN: Unpublished tracing.) |
| At least two mechanisms underlie the vagal effects on ventricular myocardium. First, the ACh released from vagus nerve endings can interact with muscarinic receptors in the cardiac cell membrane (Fig. 18-19). This interaction inhibits adenylyl cyclase, which consequently diminishes [cAMP]i and thus decreases the cAMP-dependent increase in contractility. Second, the ACh released from vagal endings can also inhibit the release of norepinephrine from neighboring sympathetic
nerve endings (Fig. 18-19). Thus, vagal activity can decrease ventricular contractility partly by antagonizing any stimulatory effects that concomitant sympathetic activity may be exerting on ventricular contractility. Similarly, sympathetic nerves release norepinephrine and certain neuropeptides, including neuropeptide Y (NPY). NPY inhibits the release of ACh from neighboring vagal fibers (Fig. 18-19).
|
| page 381 | | | page 382 |
| The adrenal medulla is essentially a component of the autonomic nervous system (see Chapters 11 and 42). The principal hormone secreted by the adrenal medulla is epinephrine; some norepinephrine is also released. The rate of secretion of these catecholamines by the adrenal
medulla is regulated by mechanisms that control the activity of the sympathetic nervous system. Thus, concentrations of catecholamines in blood rise under the same conditions that activate the sympathetic nervous system. However, the cardiovascular effects of circulating catecholamines are probably minimal under normal conditions. Moreover, the pronounced changes in myocardial contractility seen with exercise, for example, are mediated mainly by the norepinephrine released from cardiac sympathetic nerve fibers rather than by the catecholamines released from the adrenal medulla.
|
| How adrenocortical steroids influence myocardial contractility is controversial. Cardiac muscle taken from adrenalectomized animals and placed in a tissue bath is more likely to fatigue in response to stimulation than is cardiac muscle obtained from normal animals. In some species, however, adrenocortical hormones enhance contractility. In addition, the glucocorticoid hydrocortisone potentiates the cardiotonic effects of catecholamines. This potentiation is mediated in part by the ability of adrenocortical steroids to inhibit the extraneuronal catecholamine uptake mechanisms.
|
| Cardiovascular problems are common in adrenocortical insufficiency (Addison's disease). Blood volume tends to fall, which may lead to severe hypotension and cardiovascular collapse, the so-called addisonian crisis (see Chapter 42). |
| Thyroid hormone exerts its cardiac actions by two paths, genomic and nongenomic. The genomic route involves interaction of thyroxine (T3) with nuclear receptors that regulate the transcription of T3-responsive genes. In hyperthyroidism, there is increased mRNA for cardiac myocyte proteins involved in regulating [Ca++]i (SERCA, ryanodine channel) and contractile proteins (myosin heavy chain, actin, troponin I). Consequently, the rates of contraction and relaxation increase as ATP hydrolysis and O2 consumption do. There is less efficient use of ATP and greater fractional loss of heat in the hyperthyroid state. If untreated, severe hyperthyroidism can result in heart failure. |
|
| Cardiac activity is depressed in patients with inadequate thyroid function (hypothyroidism). The converse is true in patients with overactive thyroid glands (hyperthyroidism). Characteristically, hyperthyroid patients exhibit tachycardia, high cardiac output, and arrhythmias such as atrial fibrillation. In hyperthyroid subjects, sympathetic neural activity may be increased, or the sensitivity of the heart to such activity may be enhanced. Studies have shown that thyroid hormone increases the density of β-adrenergic receptors in cardiac tissue (see also Chapter 41). In experimental animals, the cardiovascular manifestations of hyperthyroidism may be simulated by the administration of excess thyroxine. |
|
| Thyroid hormones enhance myocardial contractility. Rates of ATP hydrolysis and Ca++ uptake by the sarcoplasmic reticulum are increased in experimental hyperthyroidism; the opposite effects occur in hypothyroidism. Thyroid hormones increase cardiac protein synthesis, and this response leads to cardiac hypertrophy. These hormones also affect the composition of myosin isoenzymes
in cardiac muscle. By increasing isoenzymes with the greatest ATPase activity, thyroid hormones enhance myocardial contractility.
|
| The cardiovascular changes in thyroid dysfunction also depend on indirect mechanisms. Thyroid hyperactivity increases the body's metabolic rate, which in turn results in arteriolar vasodilation. The consequent reduction in total peripheral resistance increases cardiac output, as explained in Chapter 19.
|
| Insulin has a positive inotropic effect on the heart. The effect of insulin is evident even when hypoglycemia is prevented by glucose infusions and when β-adrenergic receptors are blocked. Indeed, the positive inotropic effect of insulin is potentiated by β-adrenergic receptor antagonists. The enhanced contractility cannot be explained satisfactorily by the concomitant augmentation of glucose transport into myocardial cells.
|
| Glucagon has potent positive inotropic and chronotropic effects on the heart. This endogenous hormone is probably not important in normal regulation of the cardiovascular system, but it has been used clinically to enhance cardiac performance. The effects of glucagon on the heart and certain metabolic effects are similar to those of catecholamines. Both glucagon and catecholamines activate adenylyl cyclase to increase myocardial levels of cAMP. The catecholamines activate adenylyl cyclase by interacting with β-adrenergic receptors, but glucagon activates this enzyme by a different mechanism. Nevertheless, the rise in cAMP increases influx of Ca++ through Ca++ channels in the sarcolemma and facilitates release and reuptake of Ca++ by the sarcoplasmic reticulum, just as catecholamines do.
|
| Anterior Pituitary Hormones
|
| page 382 | | | page 383 |
| The cardiovascular derangements in hypopituitarism are related principally to the associated deficiencies in adrenocortical and thyroid function. Growth hormone affects the myocardium, at least in combination with thyroxine. In hypophysectomized animals, growth hormone alone has little effect on the depressed heart, whereas thyroxine by itself restores adequate cardiac performance
under basal conditions. However, when blood volume or peripheral resistance is increased, thyroxine alone does not restore adequate cardiac function, but the combination of growth hormone and thyroxine does reestablish normal cardiac performance. In certain animal models of heart failure, administration of growth hormone alone increases cardiac output and myocardial contractility.
|
| Changes in cardiac performance as a result of stimulation of central and peripheral chemoreceptors have been described. These effects usually predominate. However, direct effects of O2 and CO2 on the myocardium do occur.
|
| Hypoxia has a biphasic effect on myocardial performance. Mild hypoxia stimulates performance, but more severe hypoxia depresses performance because oxidative metabolism is limited.
|
| Carbon Dioxide and Acidosis
|
| An increase in Pco2 (↓pH) has a direct depressant effect on the heart. This effect is mediated by changes in intracellular pH.
|
| A reduction in intracellular pH, induced by an increase in Pco2, diminishes the amount of Ca++ released from the sarcoplasmic reticulum in response to excitation. The diminished pH also decreases the sensitivity of the myofilaments to Ca++. Increases in intracellular pH have the opposite effect; that is, they enhance sensitivity to Ca++.
|
| REGULATION OF THE PERIPHERAL CIRCULATION
|
| The peripheral circulation is essentially under dual control: centrally through the nervous system and locally by conditions in tissues surrounding the blood vessels. The relative importance of these two control mechanisms varies in different tissues (see Chapter 17).
|
| The arterioles are involved in regulating the rate of blood flow throughout the body. These vessels offer the greatest resistance to the flow of blood pumped to the tissues by the heart, and thus these vessels are important in the maintenance of arterial blood pressure. The walls of these resistance vessels are composed in large part of smooth muscle fibers that allow the diameter of the vessel lumen to vary. When this smooth muscle contracts strongly, the endothelial lining folds inward and completely obliterates the vessel lumen. When the smooth muscle is completely relaxed, the vessel lumen is maximally dilated. Some resistance vessels are closed at any given time. In addition, the smooth muscle in these vessels is partially contracted (which accounts for the tone of these vessels). If all the resistance vessels in the body dilated simultaneously, arterial blood pressure would fall precipitously.
|
|
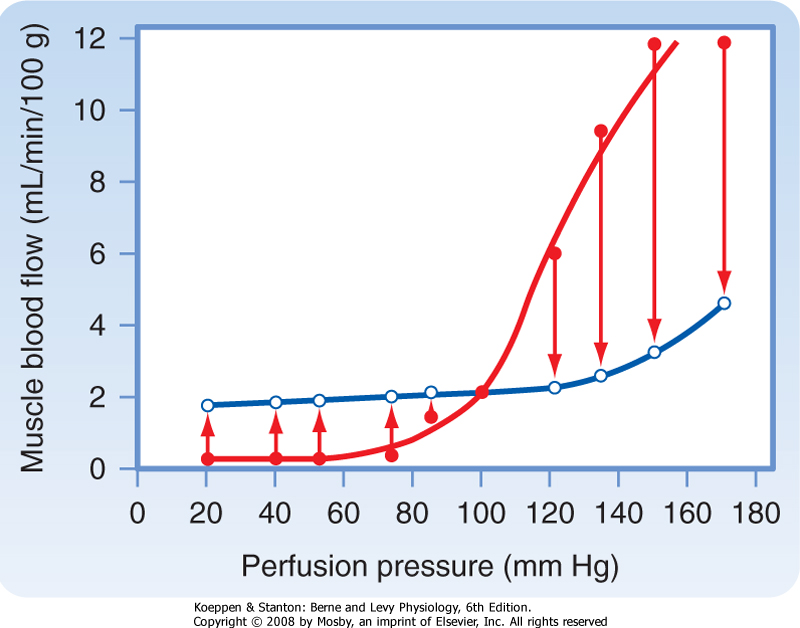
| Figure 18-22 Pressure-flow relationship in the skeletal muscle vascular bed. Closed circles represent the flows obtained immediately after abrupt changes in perfusion pressure from the control level (point where lines cross). Open circles represent the steady-state flows obtained at the new perfusion pressure. (Redrawn from Jones RD, Berne RM: Circ Res 14:126, 1964.) |
| Vascular smooth muscle controls total peripheral resistance, arterial and venous tone, and the distribution of blood flow throughout the body. The properties of vascular smooth muscle are discussed in Chapter 14.
In the following sections, intrinsic and extrinsic control of vascular smooth muscle tone, and thus perfusion of peripheral tissues, is reviewed.
|
| Intrinsic or Local Control of Peripheral Blood Flow
|
| Autoregulation and Myogenic Regulation
|
| In certain tissues, blood flow is adjusted to the existing metabolic activity of the tissue. Furthermore, when tissue metabolism is steady, changes in perfusion pressure (arterial blood pressure) evoke changes in vascular resistance that tend to maintain a constant blood flow. This mechanism, which is illustrated graphically in Figure 18-22, is commonly referred to as autoregulation of blood flow. When pressure is abruptly increased or decreased from a control pressure of 100 mm Hg, flow increases or decreases, respectively. However, even with pressure maintained at its new level, blood flow returns toward the control level within 30 to 60 seconds. Over the pressure range of 20 to 120 mm Hg, the steady-state flow is relatively constant. Calculation of hydraulic resistance (pressure/flow) across the vascular bed during steady-state conditions shows that the resistance vessels constrict with an elevation in perfusion pressure but dilate with a reduction in perfusion pressure. This response to perfusion pressure is independent of the endothelium because it is identical in intact vessels and in vessels that have been stripped of their endothelium. According to the myogenic mechanism, vascular smooth muscle contracts in response to an increase in the pressure difference across the wall of a blood vessel (transmural pressure), and it relaxes in response to a decrease in transmural pressure. The signaling mechanisms that allow distention of a vessel to elicit contraction are unknown. However, because stretch of vascular smooth muscle has been shown to raise [Ca++]i, an increase in transmural pressure is believed to activate membrane Ca++ channels.
|
| page 383 | | | page 384 |
| Transient receptor potential (TRP) channels have been implicated in the myogenic mechanism. These channels are mammalian homologues of a Drosophila melanogaster gene that when mutated, allows only a transient response to a sustained light stimulus. The pressure-induced vasoconstrictive response of an artery (myogenic response) appears to have the following signal path: pressure → increased phospholipase C activity → synthesis of diacylglycerol → activation of TRP channel → smooth muscle depolarization and opening of L-type Ca++ channels that increase [Ca++]i and muscle tone. This is a means to regulate vascular resistance. Other TRP channel types have been proposed to participate in chronic hypoxic pulmonary hypertension and in the vasoconstriction caused by the α-adrenergic agonist norepinephrine. |
|
| In normal subjects, blood pressure is maintained at a fairly constant level via the baroreceptor reflex. Hence, the myogenic mechanism may play little role in regulating blood flow to tissues under normal conditions. However, when a person changes from a lying to a standing position, transmural pressure rises in the lower extremities, and the precapillary vessels constrict in response to this imposed stretch.
|
| Endothelium-Mediated Regulation
|
| As described in Chapter 17, the endothelium lining the vasculature produces a number of substances that can relax (e.g., nitric oxide) or contract (e.g., angiotensin II and endothelin) vascular smooth muscle. Thus, the endothelium can play an important role in regulating blood flow to specific vascular beds.
|
| The metabolic activity of a tissue governs blood flow in that tissue. Any intervention that results in an inadequate O2 supply prompts the formation of vasodilator metabolites that are released from the tissue and act locally to dilate the resistance vessels. When the metabolic rate of the tissue increases or O2 delivery to the tissue decreases, more vasodilator substances are released (see Chapter 17).
|
| Candidate Vasodilator Substances
|
| Many substances have been proposed as mediators of metabolic vasodilation. Some of the earliest vasodilators suggested were lactic acid, CO2, and H+. However, the decrease in vascular resistance caused by supernormal concentrations of these vasodilators is much less than the dilation seen when metabolic activity is increased physiologically.
|
| Alterations in Po2 can change the contractile state of vascular smooth muscle. An increase in Po2 elicits contraction; a decrease elicits relaxation. However, measurements of Po2 in resistance vessels indicate that over a wide range of Po2 values (11 to 343 mm Hg), Po2 and arteriolar diameter are not well correlated. Hence, the observed changes in arteriolar diameter are more compatible with the release of a vasodilator metabolite from the tissue than with a direct effect of Po2 on vascular smooth muscle.
|
| Potassium ions, inorganic phosphate ions, and interstitial fluid osmolarity can also induce vasodilation. Both K+ and phosphate are released and osmolarity is increased during skeletal muscle contraction. Therefore, these factors may contribute to active hyperemia (increased blood flow caused by enhanced tissue activity). However, significant increases in the phosphate concentration and in osmolarity are not always observed during muscle contraction, and they may increase blood flow only transiently. Therefore, they probably do not mediate the vasodilation observed during muscular activity. Potassium is released at the onset of skeletal muscle contraction or with an increase in cardiac muscle activity. Hence, release of K+ could underlie the initial decrease in vascular resistance observed in response to physical exercise or to increased cardiac work. However, release of K+ is not sustained, yet continued arteriolar dilation persists throughout the period of enhanced muscle activity. Furthermore, reoxygenated venous blood obtained from active cardiac and skeletal muscles does not elicit vasodilation when the blood is infused into a test vascular bed. It is unlikely that oxygenation of venous blood alters its K+ or phosphate content or its osmolarity and thereby neutralizes its vasodilator effect. Therefore, some agent other than K+ must mediate the vasodilation associated with metabolic activity of the tissue.
|
| Adenosine, which contributes to the regulation of coronary blood flow, may also participate in control of the resistance vessels in skeletal muscle. In addition, some prostaglandins may be important vasodilator mediators in certain vascular beds. Thus, many candidates have been proposed as mediators of metabolic vasodilation, and the relative contribution of each remains to be determined.
|
| Metabolic control of vascular resistance by the release of a vasodilator substance requires the existence of a basal vessel tone. Tonic activity in vascular smooth muscle is readily demonstrable, but in contrast to tone in skeletal muscle, the tone in vascular smooth muscle is independent of the nervous system. Thus, some metabolic factor must be responsible for maintaining this tone. The following factors may be involved: (1) the myogenic response to the stretch imposed by blood pressure, (2) the high Po2 of arterial blood, or (3) the presence of Ca++.
|
| If arterial inflow to a vascular bed is stopped temporarily, blood flow on release of the occlusion immediately exceeds the flow that prevailed before occlusion, and the flow gradually returns to the control level. This increase in blood flow is called reactive hyperemia. This type of experiment provides evidence for the existence of a local metabolic factor that regulates tissue blood flow.
|
| page 384 | | | page 385 |
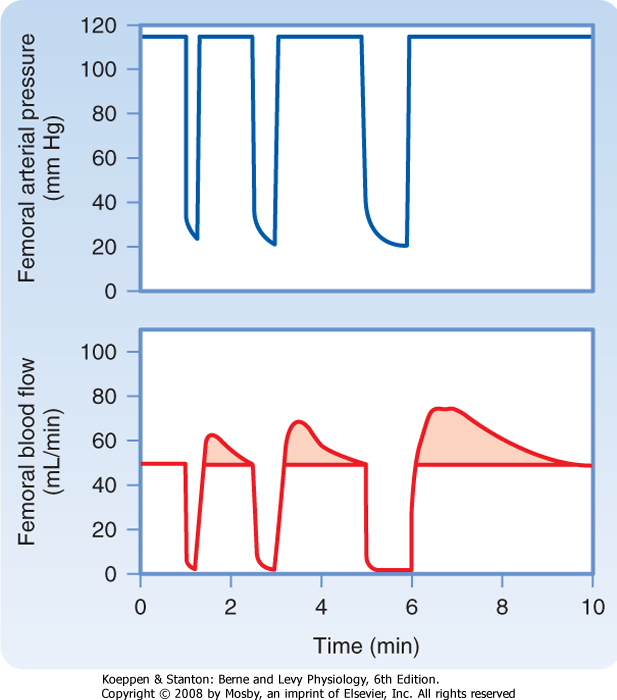
| Figure 18-23 Reactive hyperemia in the hind limb of the leg after 15-, 30-, and 60-second occlusion of the femoral artery. (From Berne RM: Unpublished observations.) |
| In the experiment shown in Figure 18-23, blood flow to the leg was stopped by clamping the femoral artery for 15, 30, and 60 seconds. Release of the 60-second
occlusion resulted in a peak blood flow that was 70% greater than the control flow, and the flow returned to the control level within 110 seconds.
|
| Within limits, peak flow and particularly the duration of reactive hyperemia are proportional to the duration of the occlusion (Fig. 18-23). If the extremity is exercised during the occlusion period, reactive hyperemia is increased. These observations and the close relationship that exists between metabolic activity and blood flow in an unoccluded limb are consistent with a metabolic mechanism in the local regulation of tissue blood flow.
|
| Coordination of Arterial and Arteriolar Dilation
|
| When the vascular smooth muscle of arterioles relaxes in response to vasodilator metabolites whose release is caused by a decrease in the ratio of O2 supply to O2 demand of the tissue, resistance may diminish concomitantly in the small upstream arteries that feed these arterioles. The result is a blood flow greater than that produced by arteriolar dilation alone. There are two possible mechanisms for this coordination of arterial and arteriolar dilation. First, the vasodilation in the microvessels may be propagated, and when dilation is initiated in the arterioles, it can propagate along the vessels from the arterioles back to the small arteries. Second, the metabolite-mediated dilation of the arterioles accelerates blood flow in the feeder arteries. This greater blood flow velocity increases the shear stress on the arterial endothelium, which in turn can induce vasodilation by release of nitric oxide.
|
| Disease of the arterial walls can lead to obstruction of the arteries, and symptoms, called intermittent claudication, appear when the arterial disease occurs in the legs. The symptoms consist of leg pain when the subject walks or climbs stairs, and the pain is relieved by rest. The disease is called thromboangitis obliterans, and it appears most frequently in men who are smokers. With minimal walking, the resistance vessels become maximally dilated by local release of metabolites; when the O2 demand of the muscles increases with more rapid walking, blood flow cannot increase sufficiently to meet the muscle needs for O2, and pain caused by muscle ischemia results. |
| Extrinsic Control of Peripheral Blood Flow
|
| Sympathetic Neural Vasoconstriction
|
| Several regions in the cerebral medulla influence cardiovascular activity. Stimulation of the dorsal lateral medulla (pressor region) evokes vasoconstriction, cardiac acceleration, and enhanced myocardial contractility. Stimulation of cerebral centers caudal and ventromedial to the pressor region decreases arterial blood pressure. This depressor area exerts its effect by direct inhibition of spinal regions and by inhibition of the medullary pressor region. These areas are not true anatomical centers in which a discrete group of cells is discernible, but they constitute "physiological" centers.
|
| The cerebrospinal vasoconstrictor regions are tonically active. Reflexes or humoral stimuli that enhance this activity increase the frequency of impulses that reach the terminal neural branches to the vessels. A constrictor neurohumor (norepinephrine) is released at the terminals to elicit a constrictive α-adrenergic effect on the resistance vessels. Inhibition of the vasoconstrictor areas diminishes the impulse frequency in the efferent nerve fibers, and vasodilation results. Thus, neural regulation of the peripheral circulation is achieved mainly by altering the impulse frequency in the sympathetic nerves to the blood vessels. Surgical section of the sympathetic nerves to an extremity abolishes sympathetic vascular tone and thereby increases blood flow to that limb. With time, vascular tone is regained by an increase in basal (intrinsic) tone.
|
| Both the pressor and depressor regions may undergo rhythmic changes in tonic activity that are manifested as oscillations in arterial pressure. Some rhythmic changes (Traube-Hering waves) occur at the frequency of respiration and are caused by a cyclic fluctuation in sympathetic impulses to the resistance vessels. Other fluctuations in sympathetic activity (Mayer waves) occur at a frequency lower than that of respiration.
|
| page 385 | | | page 386 |
| Sympathetic Constrictor Influence on Resistance and Capacitance Vessels
|
| Vasoconstrictor fibers of the sympathetic nervous system supply the arteries, arterioles, and veins; the neural influence on larger vessels is much less than it is on arterioles and small arteries. Capacitance vessels (veins) respond more to sympathetic nerve stimulation than resistance vessels do; the capacitance vessels are maximally constricted at a lower stimulation frequency than the resistance vessels are. However, capacitance vessels lack β-adrenergic receptors, and they do respond less to vasodilator metabolites. Norepinephrine is the neurotransmitter released at the sympathetic nerve terminals in blood vessel. Factors such as circulating hormones and particularly locally released substances mediate the release of norepinephrine from the nerve terminals.
|
|
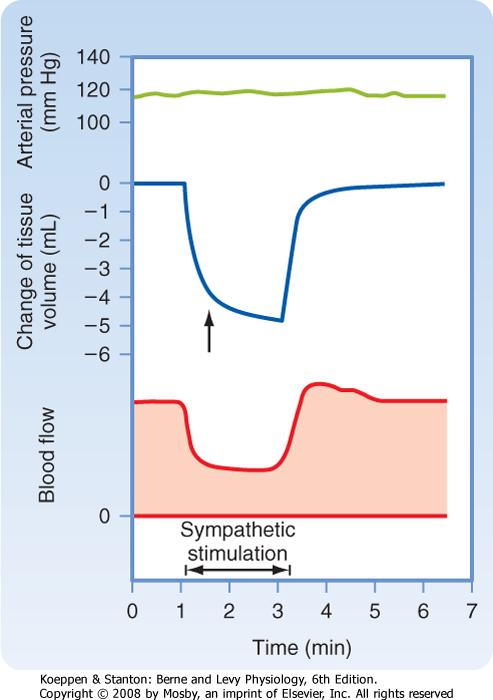
| Figure 18-24 Effect of sympathetic nerve stimulation (2 Hz) on blood flow and tissue volume of the lower limb. The arrow denotes the change in slope of the tissue volume curve at the point where the decrease in volume caused by emptying of capacitance vessels ceases and loss of extravascular fluid becomes evident. The abrupt decrease in tissue volume is caused by movement of blood out of the capacitance vessels and out of the lower limb. The late, slow progressive decline in volume (to the right of the arrow) was caused by extravascular fluid moving into the capillaries and hence away from the tissue. The loss of tissue fluid results from the lowered capillary hydrostatic pressure secondary to constriction of the resistance vessels. (From Mellander S: Acta Physiol Scand Suppl 50[176]:1, 1960.) |
| The response of the resistance and capacitance vessels to stimulation of sympathetic fibers is illustrated in Figure 18-24. When arterial pressure is held constant, stimulation of sympathetic fibers reduces blood flow (constriction of resistance vessels) and decreases the blood volume of the tissue (constriction
of capacitance vessels). Constriction of the resistance vessels established a new equilibrium of the forces responsible for filtration and absorption across the capillary wall (see Chapter 17).
|
| In addition to active changes (contraction and relaxation of vascular smooth muscle) in vessel caliber, passive changes are also caused by alterations in intraluminal pressure. An increase in intraluminal pressure distends the vessels, and a decrease reduces the caliber of the vessels as a consequence of elastic recoil of the vessel walls.
|
| At basal vascular tone, approximately a third of the blood volume of a tissue can be mobilized when the sympathetic nerves are stimulated at physiological frequencies. Basal tone is very low in capacitance vessels; if these vessels are denervated experimentally, the increases in volume evoked by maximal doses of ACh are small. Therefore, at basal vascular tone, blood volume is close to the maximal blood volume of the tissue. More blood can be mobilized from the capacitance vessels in the skin than from those in the muscle. This disparity depends in part on the greater sensitivity of the skin vessels to sympathetic stimulation, but also in part because basal tone is lower in skin vessels than in muscle vessels. Therefore, in the absence of a neural influence, skin capacitance vessels contain more blood than muscle capacitance vessels do.
|
| Physiological stimuli mobilize blood from capacitance vessels. For example, during physical exercise, activation of sympathetic nerve fibers constricts the peripheral veins and hence augments cardiac filling pressure. In arterial hypotension (as in hemorrhage), the capacitance vessels constrict and thereby correct the decreased central venous pressure associated with blood loss.
|
| Parasympathetic Neural Influence
|
| In hemorrhagic shock, the resistance vessels constrict and thereby assist in the maintenance of normal arterial blood pressure. With arterial hypotension, the enhanced arteriolar constriction also leads to a small mobilization of blood from the tissue by virtue of recoil of the postarteriolar vessels when intraluminal pressure is reduced. Furthermore, extravascular fluid is mobilized because of greater fluid absorption into the capillaries in response to the lowered capillary hydrostatic pressure (see also Chapter 19). |
| page 386 | | | page 387 |
| The efferent fibers of the cranial division of the parasympathetic nervous system innervate the blood vessels of the head and some of the viscera, whereas fibers of the sacral division innervate blood vessels of the genitalia, bladder, and large bowel. Skeletal muscle and skin do not receive parasympathetic innervation. The effect of cholinergic fibers on total vascular resistance is small because only a small proportion of the
resistance vessels of the body receive parasympathetic fibers.
|
| Stimulation of the parasympathetic fibers to the salivary glands induces marked vasodilation. A vasodilator polypeptide, bradykinin, formed locally from the action of an enzyme on a plasma protein substrate in the glandular lymphatics mediates this vasodilation. Bradykinin is formed in other exocrine glands, such as the lacrimal and sweat glands. Its presence in sweat may be partly responsible for the dilation of cutaneous blood vessels.
|
| Epinephrine and norepinephrine exert a powerful effect on peripheral blood vessels. In skeletal muscle, low concentrations of epinephrine dilate resistance vessels (β-adrenergic effect), but high concentrations produce constriction (α-adrenergic effect). In all vascular beds the primary effect of norepinephrine is vasoconstriction. When stimulated, the adrenal gland can release epinephrine and norepinephrine into the systemic circulation. However, under physiological conditions, the effect of catecholamine release from the adrenal medulla is less important than norepinephrine release from sympathetic nerve endings.
|
| Areas of the cerebral medulla that mediate sympathetic and vagal effects are under the influence of neural impulses that originate in the baroreceptors, chemoreceptors, hypothalamus, cerebral cortex, and skin. These areas of the medulla are also affected by changes in the blood concentrations of CO2 and O2.
|
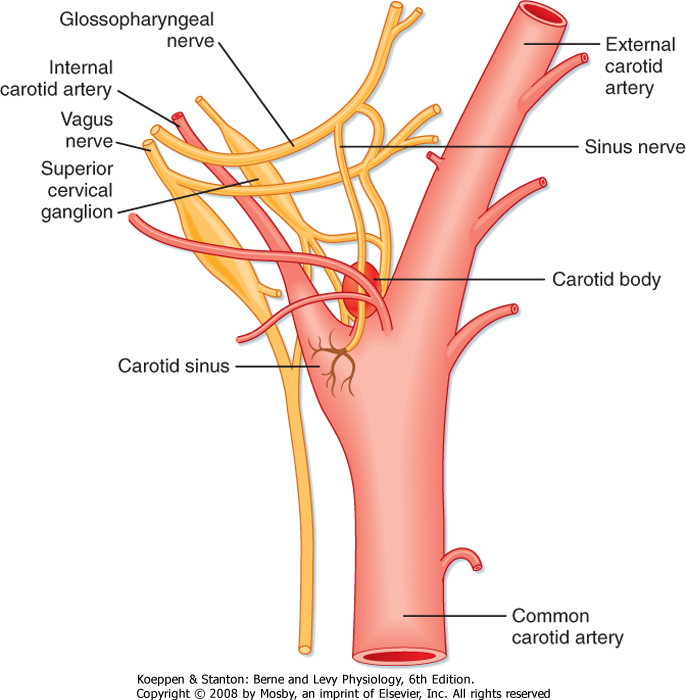
| The baroreceptors (or pressoreceptors) are stretch receptors located in the carotid sinuses and in the aortic arch (Figs. 18-25 and 18-26). The carotid sinuses are the slightly widened areas at the origins of the internal carotid arteries. Impulses that arise in the carotid sinus travel up the carotid sinus nerve (nerve of Hering) to the glossopharyngeal nerve (cranial nerve IX) and, via the latter, to the nucleus of the tractus solitarius (NTS) in the medulla. The NTS is the site of the central projections of the chemoreceptors and baroreceptors. Stimulation of the NTS inhibits sympathetic nerve outflow to the peripheral blood vessels (depressor effect), whereas lesions of the NTS produce vasoconstriction (pressor effect). Impulses that arise in the aortic arch baroreceptors reach the NTS via afferent fibers in the vagus nerves.
|
| Baroreceptor nerve terminals in the walls of the carotid sinus and aortic arch respond to the vascular stretch and deformation induced by changes in arterial blood pressure. The frequency of firing of these nerves is enhanced by an increase in arterial blood pressure and diminished by a reduction in arterial blood pressure. An increase in impulse frequency, as occurs with a rise in arterial pressure, inhibits the cerebral vasoconstrictor regions and results in peripheral vasodilation and lowering of arterial blood pressure. Bradycardia brought about by activation of the cardiac branches of the vagus nerves contributes to this lowering of blood pressure.
|
| The carotid sinus baroreceptors are more sensitive than those in the aortic arch. Changes in carotid sinus pressure evoke greater changes in systemic arterial pressure and peripheral resistance than do equivalent changes in aortic arch pressure.
|
| Figure 18-25 Diagrammatic representation of the carotid sinus and carotid body and their innervation. (Redrawn from Adams WE: The Comparative Morphology of the Carotid Body and Carotid Sinus. Springfield, IL, Charles C Thomas, 1958.) |
| page 387 | | | page 388 |
| Figure 18-26 Anterior view of the aortic arch showing the innervation of the aortic bodies and baroreceptors. (Modified from Nonidez JF: Anat Rec 69:299, 1937.) |
| The receptors in the carotid sinus walls respond more to pulsatile pressure than to constant pressure.
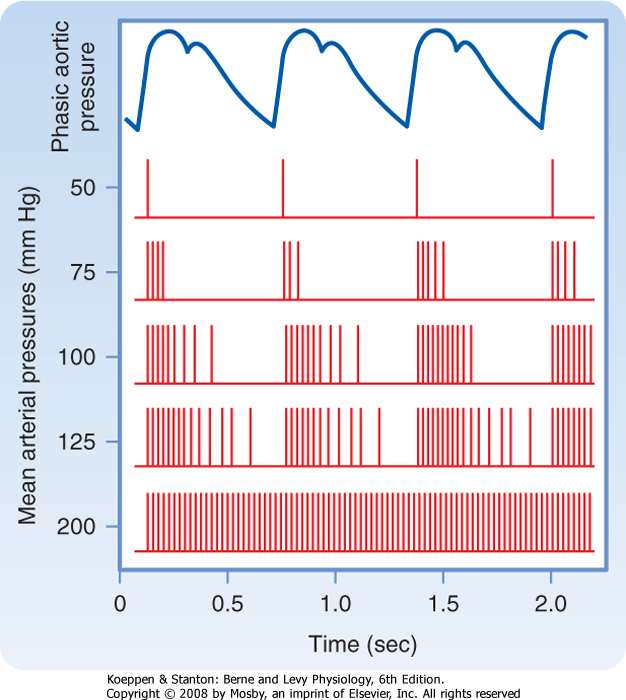
This is illustrated in Figure 18-27, which shows that at normal levels of mean arterial blood pressure (about 100 mm Hg), a barrage of impulses from a single fiber of the sinus nerve is initiated in early systole by the pressure rise; only a few spikes occurred during late systole and early diastole. At lower arterial pressure, these phasic changes are even more evident, but the overall discharge frequency is reduced. The blood pressure threshold for evoking sinus nerve impulses is about 50 mm Hg; maximal sustained firing is reached at around 200 mm Hg. Because the baroreceptors adapt, their response at any mean arterial pressure level is greater to a large than to a small pulse pressure.
|
| The increases in resistance that occur in response to reduced pressure in the carotid sinus vary from one peripheral vascular bed to another. These variations allow blood flow to be redistributed. The resistance changes elicited by altering carotid sinus pressure are greatest in the femoral vessels, less in the renal vessels, and least in the mesenteric and celiac vessels.
|
| Figure 18-27 Relationship of phasic aortic blood pressure in the firing of a single afferent nerve fiber from the carotid sinus at different levels of mean arterial pressure. |
| page 388 | | | page 389 |
| In addition, the sensitivity of the carotid sinus reflex can be altered. Local application of norepinephrine or stimulation of sympathetic nerve fibers to the carotid sinuses enhances the sensitivity of its receptors such that a given increase in intrasinus pressure produces a greater depressor response. Baroreceptor sensitivity decreases in hypertension because the carotid sinuses become stiffer as a result of the high intraarterial pressure. Consequently, a given increase in carotid sinus
pressure elicits a smaller decrease in systemic arterial pressure than it does at a normal level of blood pressure. Thus, the set point of the baroreceptors is raised in hypertension such that the threshold is increased and the pressure receptors are less sensitive to changes in transmural pressure. As would be expected, denervation of the carotid sinus can produce temporary and, in some instances, prolonged hypertension.
|
| The arterial baroreceptors play a key role in short-term adjustments in blood pressure in response to relatively abrupt changes in blood volume, cardiac output, or peripheral resistance (as in exercise). However, long-term control of blood pressure-over a period of days or weeks-is determined by the fluid balance of the individual, namely, the balance between fluid intake and fluid output. By far the single most important organ in the control of body fluid volume, and hence blood pressure, is the kidney (see also Chapter 34).
|
| Cardiopulmonary Baroreceptors
|
| Cardiopulmonary receptors are located in the atria, ventricles, and pulmonary vessels. These baroreceptors are innervated by vagal and sympathetic afferent nerves. Cardiopulmonary reflexes are tonically active and can alter peripheral resistance in response to changes in intracardiac, venous, or pulmonary vascular pressure.
|
| The atria contain two types of cardiopulmonary baroreceptors: those activated by the tension developed during atrial systole (A receptors) and those activated by stretch of the atria during atrial diastole (B receptors). Stimulation of these atrial receptors sends impulses up vagal fibers to the vagal center in the medulla. Consequently, sympathetic activity is decreased to the kidney and increased to the sinus node. These changes in sympathetic activity increase renal blood flow, urine flow, and heart rate.
|
| Activation of the cardiopulmonary receptors can also initiate a reflex that lowers arterial blood pressure by inhibiting the vasoconstrictor center in the cerebral medulla. Stimulation of the cardiopulmonary receptors inhibits release of angiotensin, aldosterone, and vasopressin (antidiuretic hormone); interruption of the reflex pathway has the opposite effects.
|
| In some individuals, the carotid sinus is abnormally sensitive to external pressure. Hence, tight collars or other forms of external pressure over the region of the carotid sinus may elicit marked hypotension and fainting. Such hypersensitivity is known as the carotid sinus syndrome. |
|
| The role that activation of these baroreceptors plays in the regulation of blood volume is apparent in the body's responses to hemorrhage. The reduction in blood volume (hypovolemia) enhances sympathetic
vasoconstriction in the kidney and increases the secretion of renin, angiotensin, aldosterone, and antidiuretic hormone (see also Chapter 19). The renal vasoconstriction (primarily afferent arterioles) reduces glomerular filtration and increases release of renin from the kidney. Renin acts on a plasma substrate to yield angiotensin II, which releases aldosterone from the adrenal cortex. The enhanced release of antidiuretic hormone decreases renal water excretion, and the release of aldosterone decreases renal NaCl excretion. The kidneys retain salt and water, and hence blood volume increases. Angiotensin II (formed from angiotensin I by angiotensin-converting enzyme) also raises systemic arteriolar tone.
|
| Peripheral Chemoreceptors
|
| These chemoreceptors consist of small, highly vascular bodies in the region of the aortic arch (aortic bodies, Fig. 18-26) and just medial to the carotid sinuses (carotid bodies, Fig. 18-25). These vascular bodies are sensitive to changes in the Po2, Pco2, and pH of blood. Although they primarily regulate respiration, they also influence the vasomotor regions. A reduction in arterial blood Po2 stimulates the chemoreceptors. The increased activity in afferent nerve fibers from the carotid and aortic bodies stimulates the vasoconstrictor regions and thereby increases the tone of resistance and capacitance vessels.
|
| The chemoreceptors are also stimulated by increased arterial blood Pco2 and by reduced pH. However, the reflex effect is small in comparison to the direct effects of hypercapnia (high Pco2) and acidosis on the vasomotor regions in the medulla. When hypoxia and hypercapnia occur simultaneously, the effects of the chemoreceptors are greater than the sum of the effects of each of the two stimuli when they act alone.
|
| Chemoreceptors are also located in the heart. These cardiac chemoreceptors are activated by ischemia of cardiac muscle, and they transmit the precordial pain (angina pectoris) associated with an inadequate blood supply to the myocardium.
|
| Optimal function of the cardiovascular reflexes requires integrity of the pontine and hypothalamic structures. Furthermore, these structures are responsible for behavioral and emotional control of the cardiovascular system (see also Chapter 11). Stimulation of the anterior hypothalamus produces a fall in blood pressure and bradycardia, whereas stimulation of the posterolateral region of the hypothalamus increases blood pressure and the heart rate. The hypothalamus also contains a temperature-regulating center that affects blood vessels in the skin. Stimulation by the application of cold to the skin or by cooling of the blood perfusing the hypothalamus results in constriction of the skin vessels and heat conservation, whereas warm stimuli to the skin result in cutaneous vasodilation and enhanced heat loss.
|
| page 389 | | | page 390 |
| The cerebral cortex also affects blood flow distribution in the body. Stimulation of the motor and premotor areas affects blood pressure; usually, a
pressor response occurs. However, vasodilation and depressor responses may be evoked, as in blushing or fainting, in response to an emotional stimulus.
|
| Painful stimuli can elicit either pressor or depressor responses, depending on the magnitude and location of the stimulus. Distention of the viscera often evokes a depressor response, whereas painful stimuli to the body surface generally evoke a pressor response.
|
| Inflation of the lungs initiates a reflex that induces systemic vasodilation and a decrease in arterial blood pressure. Conversely, collapse of the lungs evokes systemic vasoconstriction. Afferent fibers that mediate this reflex are in the vagus nerves and possibly also in the sympathetic nerves. Stimulation of these fibers by stretch of the lungs inhibits the vasomotor areas. The magnitude of the depressor response to lung inflation is directly related to the degree of inflation and to the existing level of vasoconstrictor tone (see also Chapter 22).
|
| Increases in Pco2 stimulate chemosensitive regions of the medulla (the central chemoreceptors), and they elicit vasoconstriction and increased peripheral resistance. A reduction in Pco2 below normal levels (in response to hyperventilation) decreases tonic activity in these areas in the medulla and thereby decreases peripheral resistance. The chemosensitive regions are also affected by changes in pH. Lowering of blood pH stimulates and a rise in blood pH inhibits these cerebral areas. These effects of changes in Pco2 and blood pH may operate through changes in cerebrospinal fluid pH, as may also the respiratory center.
|
| Po2 has little direct effect on the medullary vasomotor region. The primary effect of hypoxia is mediated by reflexes via the carotid and aortic chemoreceptors. A moderate reduction in Po2 stimulates the vasomotor region, but a severe reduction depresses vasomotor activity in the same manner by which other areas of the brain are depressed by very low O2 tension.
|
| Balance between Extrinsic and Intrinsic Factors in Regulation of Peripheral Blood Flow
|
| Cerebral ischemia, which may occur because of excessive pressure exerted by an expanding intracranial tumor, results in a marked increase in peripheral vasoconstriction. The stimulation is probably caused by local accumulation of CO2 and a reduction in O2 and possibly by excitation of intracranial baroreceptors. With prolonged, severe ischemia, central depression eventually supervenes, and blood pressure falls. |
| Dual control of peripheral vessels by intrinsic and extrinsic mechanisms evokes a number of important vascular adjustments. Such regulatory mechanisms
enable the body to direct blood flow to areas where it is most needed and away from areas that have fewer requirements. In some tissues, the effects of the extrinsic and intrinsic mechanisms are fixed; in other tissues, the ratio is changeable and depends on the state of activity of that tissue.
|
| In the brain and heart, which are vital structures with limited tolerance for a reduced blood supply, intrinsic flow-regulating mechanisms are dominant. For instance, massive discharge of the vasoconstrictor region via the sympathetic nerves, which might occur in severe, acute hemorrhage, has negligible effects on the cerebral and cardiac resistance vessels, whereas the cutaneous, renal, and splanchnic blood vessels become greatly constricted (see also Chapter 19).
|
| In the skin, extrinsic vascular control is dominant. The cutaneous vessels not only participate strongly in a general vasoconstrictor discharge but also respond selectively via hypothalamic pathways to subserve the heat loss and heat conservation functions required for regulation of body temperature. However, intrinsic control can be elicited by local temperature changes that modify or override the central influence on resistance and capacitance vessels (see also Chapter 17).
|
| In skeletal muscle, the extrinsic and intrinsic mechanisms interact. In resting skeletal muscle, neural control (vasoconstrictor tone) is dominant, as can be demonstrated by the large increase in blood flow that occurs immediately after section of the sympathetic nerves to the tissue. After the onset of exercise, the intrinsic flow-regulating mechanism assumes control, and vasodilation occurs in the active muscles because of the local increase in metabolites. Vasoconstriction occurs in the inactive tissues as a manifestation of the general sympathetic discharge. However, constrictor impulses that reach the resistance vessels of the active muscles are overridden by the local metabolic effect. Operation of this dual control mechanism thus provides increased blood flow where it is required and shunts it away from relatively inactive areas (see also Chapter 17). Similar effects may be achieved in response to an increase in Pco2. Normally, the hyperventilation associated with exercise keeps Pco2 at normal levels. However, if Pco2 is increased, generalized vasoconstriction would occur because CO2 stimulates the medullary vasoconstrictor region. In active muscles, where [CO2] would be highest, the smooth muscle of the arterioles would relax in response to the local Pco2. Factors that affect and are affected by the vasomotor region are summarized in Figure 18-28.
|
| page 390 | | | page 391 |
| page 391 | | | page 392 |
- Cardiac function is regulated by a number of intrinsic and extrinsic mechanisms. The principal intrinsic mechanisms that regulate myocardial contraction are the Frank-Starling mechanism and rate-induced regulation.
- The heart rate is regulated mainly by the autonomic nervous system. Sympathetic nervous activity increases the heart rate, whereas parasympathetic
(vagal) activity decreases the heart rate. When both systems are active, the vagal effects usually dominate. The autonomic nervous system regulates myocardial performance mainly by varying the Ca++ conductance of the cell membrane via the adenylyl cyclase system.
- The following reflexes regulate the heart rate: the baroreceptor, chemoreceptor, pulmonary inflation, atrial receptor (Bainbridge), and ventricular receptor reflexes.
- Certain hormones, such as epinephrine, adrenocortical steroids, thyroid hormones, insulin, glucagon, and anterior pituitary hormones, regulate myocardial performance. Changes in the arterial blood concentrations of O2, CO2, and H+ directly alter cardiac function and indirectly alter it via the chemoreceptors.
- The arterioles (resistance vessels) mainly regulate blood flow through their downstream capillaries. Smooth muscle, which makes up most of the walls of arterioles, contracts and relaxes in response to neural and humoral stimuli. Neural regulation of blood flow is almost completely accomplished by the sympathetic nervous system. Sympathetic nerves to blood vessels are tonically active; inhibition of the vasoconstrictor center in the medulla reduces peripheral vascular resistance. Stimulation of the sympathetic nerves constricts the resistance and capacitance (veins) vessels. Parasympathetic fibers innervate the head, viscera, and genitalia; they do not innervate the skin and muscle.
- Autoregulation of blood flow occurs in most tissues. This process is characterized by a constant blood flow in the face of a change in perfusion pressure. Autoregulation is mediated by a myogenic mechanism whereby an increase in transmural pressure elicits a contraction of vascular smooth muscle and a decrease in transmural pressure elicits a relaxation.
- The striking parallelism between tissue blood flow and tissue O2 consumption indicates that blood flow is regulated largely by a metabolic mechanism. A decrease in the O2 supply-to-O2 demand ratio of a tissue releases vasodilator metabolites that dilate arterioles and thereby enhance the O2 supply.
- The baroreceptors in the internal carotid arteries and aorta are tonically active and regulate blood pressure on a moment-to-moment basis. An increase in arterial pressure stretches these receptors to initiate a reflex that inhibits the medullary
vasoconstrictor center and induces vasodilation. Conversely, a decrease in arterial pressure disinhibits the vasoconstrictor center and induces vasoconstriction. The baroreceptors in the internal carotid arteries predominate over those in the aorta, and they respond more vigorously to changes in pressure (stretch) than they do to elevated or reduced nonpulsatile pressure.
- Peripheral chemoreceptors (carotid and aortic bodies) and central chemoreceptors in the medulla oblongata are stimulated by a decrease in blood Po2 and by an increase in blood Pco2. Stimulation of these chemoreceptors increases the rate and depth of respiration, but it also produces peripheral vasoconstriction. Cardiopulmonary baroreceptors are also present in the cardiac chambers and large pulmonary vessels. They have less influence on blood pressure but participate in regulation of blood volume.
- Peripheral resistance and hence blood pressure are affected by stimuli that arise in the skin, viscera, lungs, and brain. The combined effect of neural and local metabolic factors distributes blood to active tissues and diverts it from inactive tissues. In vital structures, such as in the heart and brain and in contracting skeletal muscle, the metabolic factors predominate.
|
|
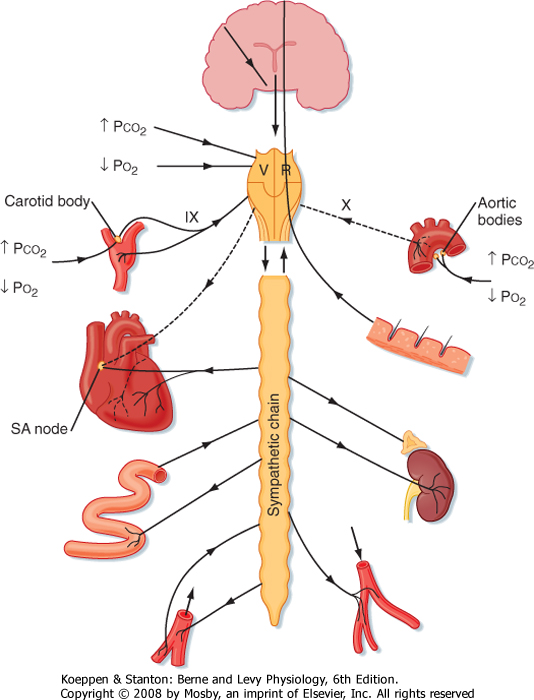
|
| Figure 18-28 Schematic diagram illustrating neural input and output of the vasomotor region (VR). IX, glossopharyngeal nerve; X, vagus nerve. |
|