| 31 Transport and Metabolic Functions of the Liver
|
| OVERVIEW OF THE LIVER AND ITS FUNCTIONS
|
| The liver is a large, multilobed organ located in the abdominal cavity whose function is intimately associated with that of the gastrointestinal system. The liver serves as the first site of processing for most absorbed nutrients and also secretes bile acids, which as we learned in Chapter 29, plays a critical role in the absorption of lipids from the diet. In addition, the liver is a metabolic powerhouse, critical for disposing of a variety of metabolic waste products and xenobiotics from the body by converting them to forms that can be excreted. The liver stores or produces numerous substances needed by the body, such as glucose, amino acids, and plasma proteins. In general, key functions of the liver can be divided into three areas: (1) contributions to whole-body metabolism, (2) detoxification, and (3) excretion of protein-bound/lipid-soluble waste products. In this chapter we discuss the structural and molecular features of the liver and the biliary system that subserve these functions, as well as their regulation. However, although the liver contributes in a pivotal way to the maintenance of whole-body biochemical status, a complete discussion of all of the underpinning reactions is beyond the scope of the present text. We will confine our discussion primarily to hepatic functions that relate to gastrointestinal physiology.
|
| Metabolic Functions of the Liver
|
| Hepatocytes contribute to metabolism of the major nutrients: carbohydrates, lipids, and proteins. Thus, the liver plays an important role in glucose metabolism by engaging in gluconeogenesis, the conversion of other sugars to glucose. The liver also stores glucose as glycogen at times of glucose excess (such as in the postprandial period) and then releases stored glucose into the bloodstream as it is needed. This process is referred to as the "glucose buffer function of the liver." When hepatic function is impaired, glucose concentrations in blood may rise excessively after the ingestion of carbohydrate; conversely, between meals, hypoglycemia may be seen because of an inability of the liver to contribute to carbohydrate metabolism and interconversion of one sugar to another.
|
| Hepatocytes also participate in lipid metabolism. They are a particularly rich source of the metabolic enzymes engaged in fatty acid oxidation to supply energy for other body functions. Hepatocytes also convert products of carbohydrate metabolism to lipids that can be stored in adipose tissue and synthesize large quantities of lipoproteins, cholesterol, and phospholipids, the latter two being important in the biogenesis of cell membranes. In addition, hepatocytes convert a considerable portion of synthesized cholesterol to bile acids, of which we will discuss more later in this chapter.
|
| The liver also plays a vital role in protein metabolism. The liver synthesizes all of the so-called nonessential amino acids (see Chapter 29) that do not need to be supplied in the diet, in addition to participating in interconverting and deaminating amino acids so that the products can enter biosynthetic pathways for the synthesis of carbohydrates. With the exception of immunoglobulins, the liver synthesizes almost all of the proteins present in plasma, especially albumin, which determines plasma oncotic pressure, as well as most of the important clotting factors. Patients suffering from liver disease may develop peripheral edema secondary to hypoalbuminemia and are also susceptible to bleeding disorders. Finally, the liver is the critical site for disposal of the ammonia generated from protein catabolism. This is accomplished by converting ammonia to urea, which can then be excreted by the kidneys. The details of this process will be discussed later.
|
| The Liver and Detoxification
|
| page 542 |  | | page 543 |
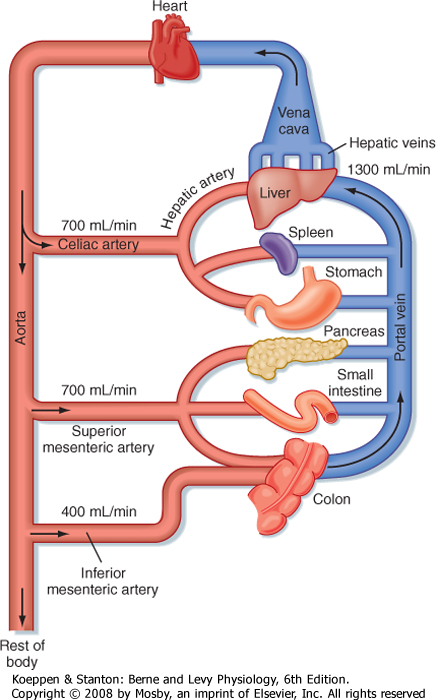
| Figure 31-1 Typical blood flow through the splanchnic circulation in a fasting adult human. |
| The liver serves both as a gatekeeper, by limiting the entry of toxic substances into the bloodstream, and as a garbage disposal, by extracting potentially toxic metabolic products produced elsewhere in the body and converting them to chemical forms that can be excreted. The liver fulfills these functions, in part, because of its unusual blood supply. Unlike all other organs, the majority of blood arriving at the liver is venous in nature and is supplied via the portal vein from the intestine (Fig. 31-1). As such, the liver is strategically located to receive not only absorbed nutrients but also potentially harmful absorbed molecules such as drugs and bacterial toxins. Depending on the efficiency with which these molecules are extracted by hepatocytes and subjected to so-called first-pass metabolism, little or none of the absorbed substance may make it into the systemic circulation. This is a major reason why not all pharmaceutical agents can
achieve therapeutic concentrations in the bloodstream if administered orally.
|
|
Table 31-1.
Key Transporters of Hepatocytes |
| Name | Basolateral | Canalicular | Substrate/Function |
| NTCP | Yes | No | Uptake of conjugated bile acids |
| OATP | Yes | No | Uptake of bile acids and xenobiotics |
| BSEP | No | Yes | Secretion of conjugated bile acids |
| MDR3 | No | Yes | Secretion of phosphatidylcholine |
| MDR1 | No | Yes | Secretion of cationic xenobiotics |
| ABC5/ABC8 | No | Yes | Secretion of cholesterol |
| cMOAT/MRP2 | No | Yes | Secretion of sulfated lithocholic acid and conjugated bilirubin |
| The liver has two levels at which it removes and metabolizes/detoxifies substances originating from the portal circulation. The first of these is physical. Blood arriving in the liver percolates among cells of macrophage lineage, known as Kupffer cells. These cells are phagocytic and are particularly important in removing particulate material from portal blood, including bacteria that may enter blood from the colon even under normal conditions. The second level of defense is biochemical. Hepatocytes are endowed with a broad array of enzymes that metabolize and modify both endogenous and exogenous toxins so that the products are, in general, more water soluble and less susceptible to reuptake by the intestine. The metabolic reactions involved are broadly divided into two classes. Phase I reactions (oxidation, hydroxylation, and other reactions catalyzed by cytochrome P-450 enzymes) are followed by phase II reactions that conjugate the resulting products with another molecule, such as glucuronic acid, sulfate, amino acids, or glutathione, to promote their excretion. The products of
these reactions are then excreted into bile or returned to the bloodstream to ultimately be excreted by the kidneys. We will return to the precise mechanisms involved in the detoxification of some key metabolic waste products later.
|
| Role of the Liver in Excretion
|
| The kidneys play an important role in the excretion of water-soluble catabolites, as discussed in the renal section. Only relatively small water-soluble catabolites can be excreted by the process of glomerular filtration. However, larger water-soluble catabolites and molecules bound to plasma proteins, including lipophilic metabolites and xenobiotics, steroid hormones, and heavy metals, cannot be filtered by the glomerulus. All these substances are potentially harmful if allowed to accumulate, so a mechanism must exist for their excretion. The mechanism for excretion involves the liver, which excretes these substances in bile. Hepatocytes take up these substances with high affinity by virtue of the presence of an array of basolateral membrane transporters, and the substances are subsequently metabolized at the level of microsomes and in the cytosol (Table 31-1). Ultimately, substances destined for excretion in bile are exported across the canalicular membrane of hepatocytes via a different array of transporters. The features of bile allow solubilization of even lipophilic substances, which can then be excreted into the intestine and ultimately leave the body in feces.
|
| STRUCTURAL FEATURES OF THE LIVER AND BILIARY SYSTEM
|
| Hepatocytes, the major cell type in the liver, are arranged in anastomosing cords that form plates around which large volumes of blood circulate (Fig. 31-2). The liver receives a high blood flow that is disproportionate to its mass, which ensures that hepatocytes receive high quantities of both O2 and nutrients. Hepatocytes receive more than 70% of their blood supply at rest via the portal vein (rising to more than 90% in the postprandial period).
|
| page 543 | | | page 544 |
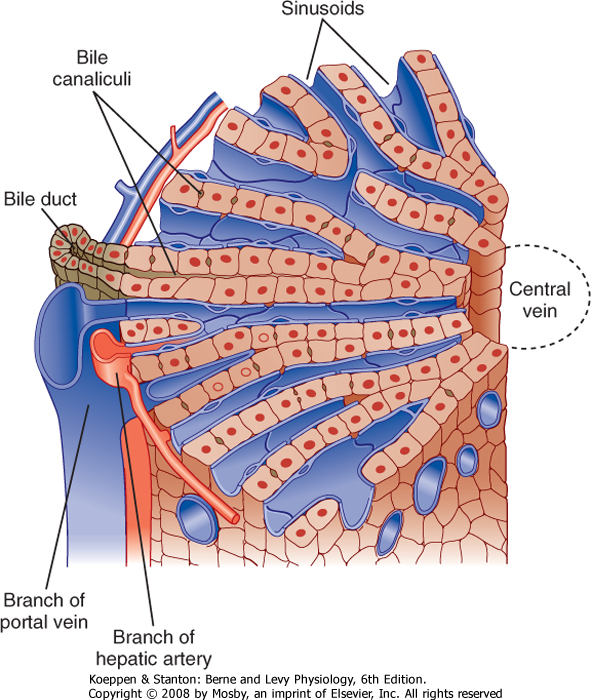
| Figure 31-2 Diagrammatic representation of a hepatic lobule. Plates of hepatocytes are arrayed radially around a central vein. Branches of the portal vein and hepatic artery are located on the periphery of the lobule and form the "portal triad" together with the bile duct. Blood from the portal vein and hepatic artery percolates around the hepatocytes via the sinusoids before draining into the central vein. (Modified from Bloom W, Fawcett DW: A Textbook of Histology, 10th ed. Philadelphia, Saunders, 1975.) |
| The plates of hepatocytes that constitute the liver parenchyma are supplied by a series of sinusoids, which are low-resistance cavities supplied by branches of both the portal vein and the hepatic artery. The sinusoids are unlike the capillaries that perfuse other organs. During fasting, many sinusoids are collapsed, but more can gradually be recruited as portal blood flow increases during the period after a meal when absorbed nutrients are transported to the liver. The low resistance of the sinusoidal cavities means that blood flow through the liver can increase considerably without a concomitant increase in pressure. Eventually, the blood drains into central branches of the hepatic vein.
|
| If the circulation of the liver, particularly its sinusoids, is compressed by fibrosis, the liver loses its ability to accommodate the increases in blood flow that occur after a meal without a concomitant increase in pressure. Because of the fenestrations, albumin escapes from the circulation and albumin-rich fluid weeps from the surface of the liver into the abdominal cavity, where it overwhelms the lymphatic drainage. This condition is known as ascites and is reflected in a considerable increase in the girth of many patients with liver disease. As pressure in the liver builds, new collateral blood vessels form in an attempt to circumvent the obstruction and reduce the portal hypertension. Some of these vessels are directed to abdominal structures and, because of their thin, weak walls, are prone to rupture. A particular example is the formation of high-pressure collaterals to the esophagus, which can then become varices that bleed into the lumen. Bleeding into the esophageal lumen is very hard to control and is thus a medical emergency. Even in the absence of bleeding, moreover, the formation of collateral blood vessels bypasses the remaining metabolic capacity of the liver, and levels of toxins such as ammonia are increased and can exert adverse effects elsewhere in the body. |
|
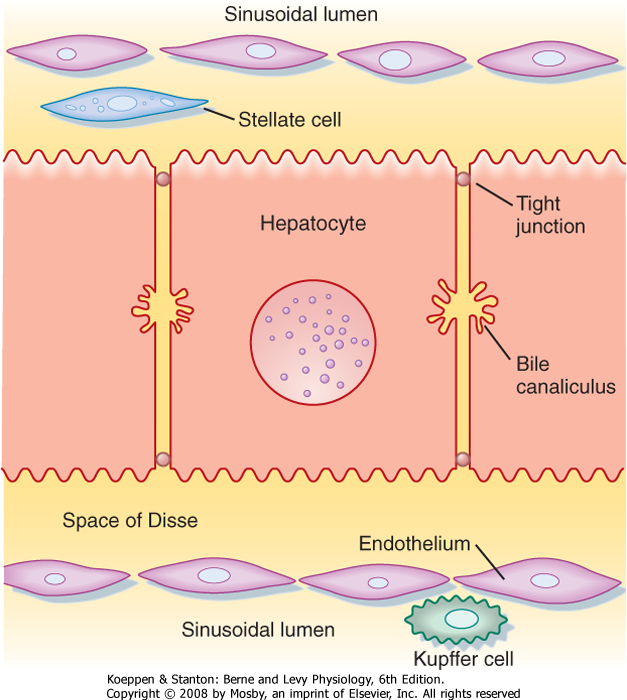
|
| Figure 31-3 Interrelationships of the major cell types in the liver. |
| page 544 | | | page 545 |
| Infection of the liver with certain viruses or overexposure to toxic substances such as alcohol kills hepatocytes and activates hepatic stellate cells, which synthesize excessive amounts of collagen that result in the histologic appearance of fibrosis. If the insult is chronic, the fibrosis eventually becomes irreversible, a condition known as cirrhosis. The fibrotic, scarred areas crowd out the hepatocyte mass, thereby reducing the synthetic, metabolic, and excretory capacity of the liver. Fibrotic masses press on the sinusoids and prevent them from expanding as portal blood flow to the liver increases during the postprandial period. Edema may develop in patients with chronic liver injury as a result of reduced levels of albumin in blood, and a condition known as ascites may then develop, in which fluid accumulates in the peritoneal cavity secondary to increased portal pressure. Eventually, the accumulation of toxic substances in the bloodstream can lead to jaundice, itching, and neurological complications. If hepatic function becomes compromised beyond a certain level, the only effective treatment is liver transplantation. |
|
| The sinusoids are also unusual in the endothelial cells that line their walls (Fig. 31-3). Hepatic endothelial cells contain specialized openings, known as fenestrations, that are large enough to permit the passage of molecules as big as albumin. Sinusoidal endothelial cells also lack a basement membrane, which might otherwise pose a diffusion barrier. These features allow access of albumin-bound substances to the hepatocytes that will eventually take them up. The sinusoids also contain Kupffer cells. Beneath the sinusoidal endothelium and separating the endothelium
from the hepatocytes is a thin layer of loose connective tissue called the space of Disse, which likewise poses little resistance to the movement of molecules even as large as albumin in health. The space of Disse is also the location of another important hepatic cell
type, the stellate cell. Stellate cells serve as storage sites for retinoids and in addition are the source of key growth factors for hepatocytes. Under abnormal conditions, stellate cells are activated to synthesize large quantities of collagen, which contributes to the hepatic dysfunction.
|
| Hepatocytes are also the origination point for the biliary system. Although hepatocytes are considered to be epithelial cells with basolateral and apical membranes, the spatial arrangement of these two cell domains differs from that seen in simple columnar epithelium, such as that lining the gastrointestinal tract. Rather, in the liver the apical surface of the hepatocyte occupies only a small fraction of the cell membrane, and the apical membranes of adjacent cells oppose each other to form a channel between the cells known as the canaliculus (Fig. 31-3). The role of canaliculi is to drain bile from the liver, and these canaliculi drain into biliary ductules, which are lined by classic columnar epithelial cells known as cholangiocytes. Ultimately, the biliary ductules drain into large bile ducts that coalesce into the right and left hepatic ducts to permit exit of bile from the liver. These, in turn, form the common hepatic duct, from which bile can flow into either the gallbladder, via the cystic duct, or the intestine, via the common bile duct (Fig. 31-4), on the basis of prevailing pressure relationships.
|
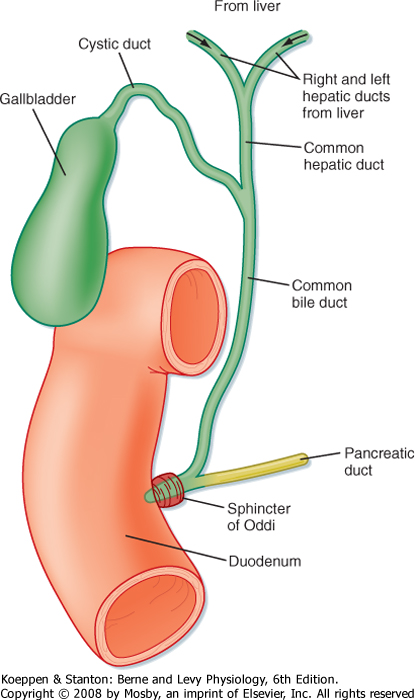
|
| Figure 31-4 Functional anatomy of the biliary system. |
| One other feature of the structural organization of the liver bears emphasis because of its clinical significance. Branches of the hepatic vein, hepatic artery, and bile ducts run in parallel in the so-called hepatic triad. Hepatocytes lying closest to this triad are referred to as periportal, or "zone 1," and have the greatest supply of oxygen and nutrients. In contrast,
hepatocytes lying closest to branches of the hepatic vein are referred to as pericentral, or "zone 3." The latter cells are more sensitive to ischemia, whereas the former are more sensitive to oxidative injury. Thus, the location of damaged cells on biopsy may provide clues to the cause of a given case of liver injury. Zone 1 cells are most active in detoxification functions in normal circumstances, but zone 2 (intermediate between zones 1 and 3) and zone 3 cells can progressively be recruited in cases of liver disease, comparable to the concept of the anatomic reserve that we considered for lipid assimilation in the small intestine. Conversely, zone 3 cells are thought to be most active in bile acid synthesis.
|
| BILE FORMATION AND SECRETION
|
| page 545 | | | page 546 |
| Bile is the excretory fluid of the liver that plays an important role in lipid digestion. Bile formation begins in hepatocytes, which actively transport solutes into bile canaliculi across their apical membranes. Bile is a micellar solution in which the major solutes are bile acids, phosphatidylcholine, and cholesterol in an approximate ratio of 10 : 3 : 1, respectively. Secretion of these solutes drives the concomitant movement of
water and electrolytes across the tight junctions that link adjacent hepatocytes to form canalicular bile. The majority of bile flow is driven by the secretion of bile acids across the apical membrane of hepatocytes via an ATPase transporter known as the bile salt export pump (BSEP; Table 31-1). The composition of the resulting fluid can be modified further as it flows through the biliary ductules (resulting in hepatic bile) and still further on storage in the gallbladder (gallbladder bile). Ultimately, bile becomes a concentrated solution of biological detergents that aids in solubilization of the products of lipid digestion in the aqueous environment of the intestinal lumen, thereby enhancing the rate at which lipids are transferred to the absorptive epithelial surface. It also serves as a medium in which metabolic waste products are exported from the body.
|
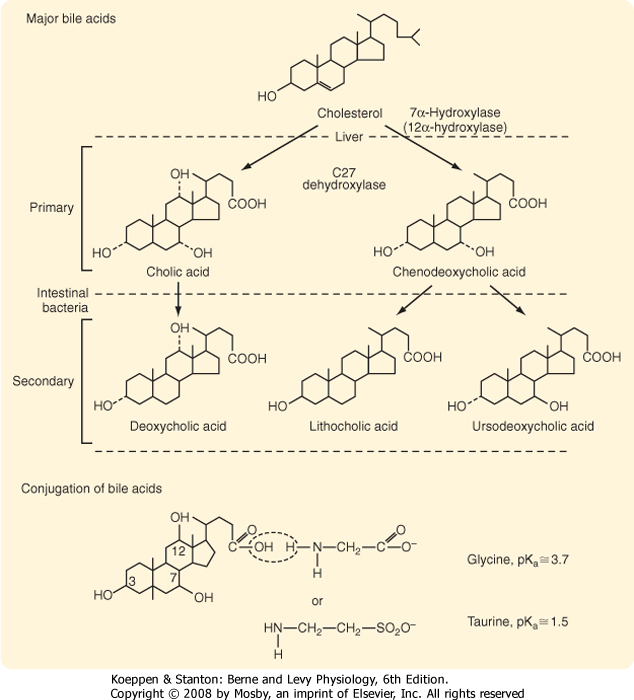
| Figure 31-5 Structures and sites of production of the major primary and secondary bile acids of bile. At the bottom of the figure, the conjugation of cholic acid with glycine or taurine is shown. |
| Though rare, a variety of familial syndromes that are manifested as progressive cholestasis have taught us a great deal about the molecular nature of the transporters that deliver bile constituents into the canaliculus. For example, type II progressive familial intrahepatic cholestasis (PFIC II) has been mapped to a mutation in BSEP, which results in an almost total absence of bile acids in bile. Cholestasis develops in patients with this disorder, but they have relatively little, if any, evidence of bile duct injury. PFIC III, on the other hand, is a much more aggressive disease in which cholestasis is accompanied by early increases in circulating γ-glutamyl transpeptidase. The molecular culprit is a mutation that abolishes expression of MDR3. In the absence of this transporter, phosphatidylcholine is no longer able to enter bile, thus illustrating the importance of this lipid in protecting cholangiocytes from the injurious effects of bile acids because mixed micelles cannot form in its absence. |
|
| page 546 | | | page 547 |
| Bile acids are produced by hepatocytes as end products of cholesterol metabolism. Cholesterol is selectively metabolized by a series of enzymes that result in the formation of bile acid (Fig. 31-5). The initial and
rate-limiting step is addition of a hydroxyl group to the 7 position of the steroid nucleus by the enzyme cholesterol 7α-hydroxylase. The side chain of the product of this reaction is then shortened and a carboxylic acid function added by C27 dehydroxylase to yield chenodeoxycholic acid, a dihydroxy bile acid. Alternatively, the product is further hydroxylated at the 12 position and then acted on by C27 dehydroxylase to yield cholic acid, a trihydroxy bile acid. Bile acid synthesis can be up- or down-regulated, depending on the body's requirements (Fig. 31-6). For example, if bile acid levels are reduced in the blood flowing to the liver, synthesis can be increased up to 10-fold. Conversely, feeding of bile acids profoundly suppresses the new synthesis of bile acids by hepatocytes. The mechanisms underlying these changes in bile acid synthesis relate to changes in expression of the enzymes involved, and bile acids have been shown to be able to directly activate specific transcription factors that mediate such regulation.
|
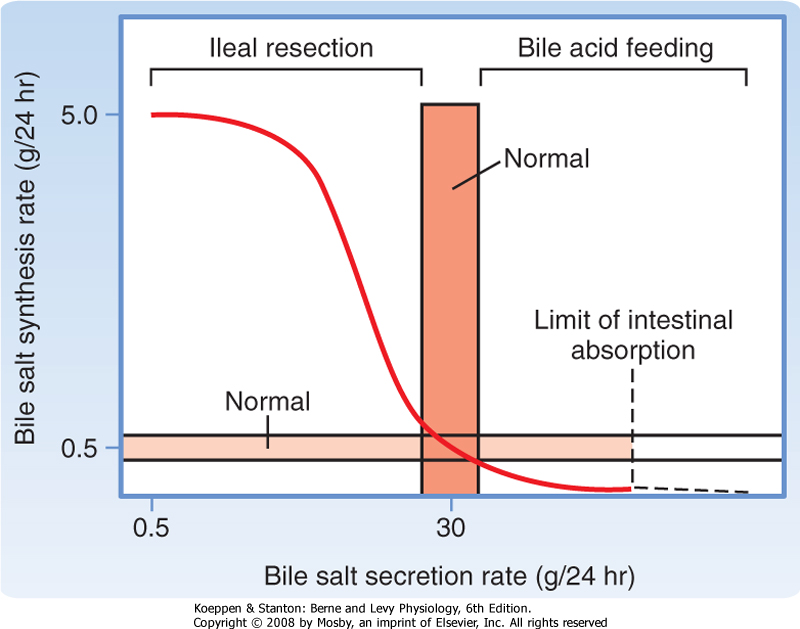
|
| Figure 31-6 Relationship between rates of bile acid synthesis and secretion. Increased secretion normally increases the rate of return of bile acids to the liver via portal blood, which exerts a negative feedback on synthesis. Conversely, interruption of the enterohepatic circulation, such as after ileal resection, can increase synthesis to values more than 10-fold higher than normal. (From Carey MC, Cahalane MJ. In Arias IM et al [eds]: The Liver: Biology and Pathobiology, 2nd ed. New York, Raven Press, 1988.) |
| Chenodeoxycholic acid and cholic acid are referred to as primary bile acids because they are synthesized by the hepatocyte (Fig. 31-5). However, each can be acted on in the colonic lumen by bacterial enzymes to yield ursodeoxycholic and deoxycholic acid, respectively. Chenodeoxycholic acid is also converted by bacterial enzymes to form lithocholic acid, which is relatively cytotoxic. Collectively, these three products of bacterial metabolism are referred to as secondary bile acids. One additional important biochemical modification occurs in both primary and secondary bile acids in the hepatocyte (Fig. 31-5). These molecules are conjugated with either glycine or taurine, which significantly depresses their pKa. The result is
that conjugated bile acids are almost totally ionized at the pH prevailing in the small intestinal lumen and thus cannot passively traverse cell membranes. Consequently, the conjugated bile acids are retained in the intestinal lumen until they are actively absorbed in the terminal ileum via the apical sodium-dependent bile salt transporter (ASBT). Conjugated bile acids that escape this uptake step are deconjugated by bacterial enzymes in the colon, and the resulting unconjugated forms are passively reabsorbed across the colonic epithelium because they are no longer charged.
|
| Hepatic Aspects of the Enterohepatic Circulation of Bile Acids
|
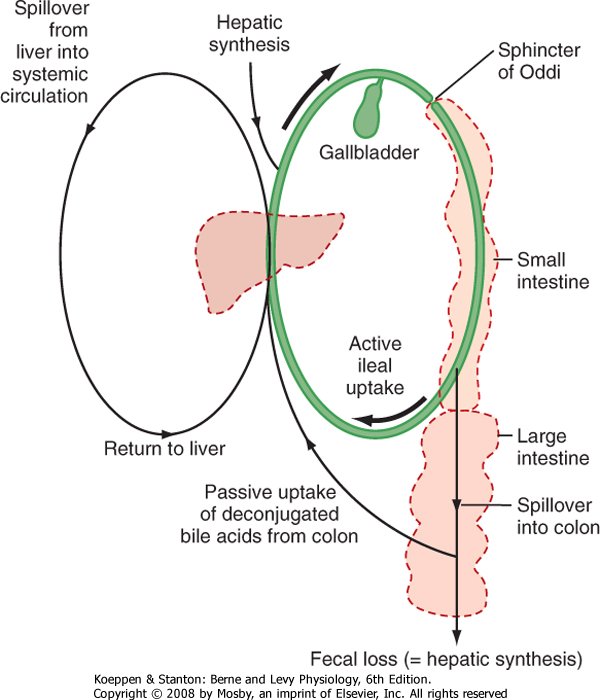
| Figure 31-7 Relative amounts of bile acids in different body pools and the enterohepatic circulation. |
| page 547 | | | page 548 |
| Bile acids assist in the digestion and absorption of lipids by acting as detergents rather than enzymes, and thus a significant mass of these molecules is required to solubilize all dietary lipids. Via the enterohepatic circulation, actively reabsorbed conjugated bile acids travel through the portal blood back to the hepatocyte, where they are efficiently taken up by basolateral transporters that may be Na+ dependent or independent (Table 31-1). Similarly, bile acids that are deconjugated in the colon also return to the hepatocyte, where they are reconjugated to be secreted into bile. In this way we acquire a pool of circulating primary and secondary bile acids, and daily synthesis is then equal only to the minor fraction (approximately 10%/day, or 200 to 400 mg) that escapes uptake and is lost in stool (Fig. 31-7). The only exception to this rule is lithocholic acid, which is preferentially sulfated in the hepatocyte rather than being conjugated with
glycine or taurine. The majority of such conjugates are lost from the body after each meal because they are not substrates for ASBT, thereby avoiding accumulation of a potentially toxic molecule.
|
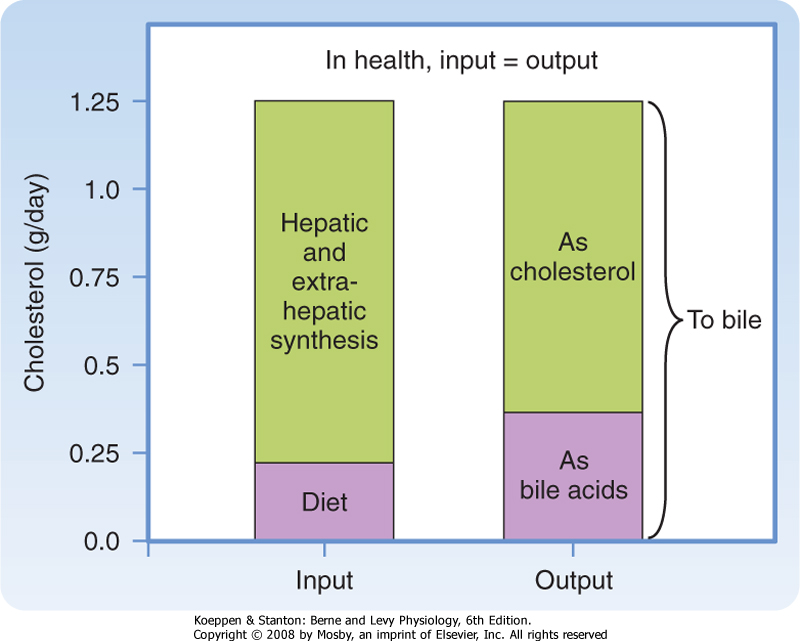
| Some comment should also be made with respect to the role of bile acids in whole-body cholesterol homeostasis. The pool of cholesterol in the body reflects its daily synthesis, as well as the relatively minor component derived from inefficient dietary uptake, balanced against loss from the body, which can only occur via bile in health (Fig. 31-8). Cholesterol can be excreted in two forms, either as the native molecule or after its conversion to bile acids. The latter account for up to a third of the cholesterol excreted per day despite enterohepatic recycling. Thus, one strategy for the treatment of hypercholesterolemia is to interrupt the enterohepatic circulation of bile acids, which drives increased conversion of cholesterol to bile acids; the bile acids are then lost from the body in feces.
|
| Figure 31-8 Daily cholesterol balance in healthy adult humans. |
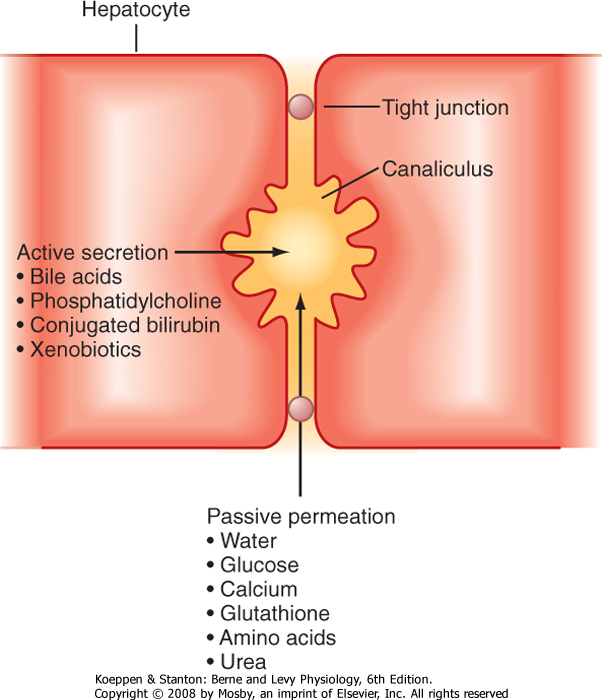
| As noted earlier, bile also contains cholesterol and phosphatidylcholine. Cholesterol transport across the canalicular membrane is mediated, at least in part, by a heterodimer of the active transporters we discussed in Chapter 29 as participating in the efflux of cholesterol from the small intestinal epithelial cells, namely, ABC5 and ABC8 (Table 31-1). Phosphatidylcholine derives from the inner leaflet of the canalicular membrane and is specifically "flipped" across the membrane by another ABC family transporter called multidrug resistance protein 3 (MDR3). Furthermore, because mixed micelles composed of bile acids, phosphatidylcholine, and cholesterol are osmotically active and the tight junctions that link adjacent hepatocytes are relatively leaky, water is drawn into the canalicular lumen, as well as other plasma solutes, such as Ca++, glucose, glutathione, amino acids, and urea, at concentrations essentially approximating those in plasma
(Fig. 31-9). Finally, conjugated bilirubin, which is water soluble, and a variety of additional organic anions and cations formed from endogenous metabolites and xenobiotics are secreted into bile across the apical membrane of the hepatocyte.
|
| Bile Modification in the Ductules
|
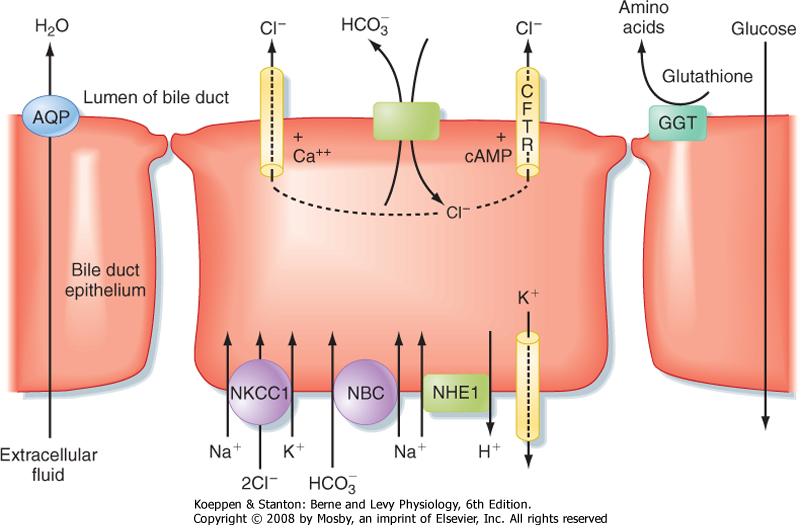
| The cholangiocytes lining the biliary ductules are specifically designed to modify the composition of bile (Fig. 31-10). Useful solutes, such as glucose and amino acids, are reclaimed by the activity of specific transporters. Chloride ions in bile are also exchanged for HCO3-, thus rendering the bile slightly alkaline and reducing the risk of precipitation of Ca++. Glutathione is broken down on the surface of cholangiocytes into its constituent amino acids by the enzyme γ-glutamyl transpeptidase (GGT), and the products are reabsorbed. The bile is also diluted at this site, in concert with ingestion of a meal, in response to hormones, such as secretin, that increase HCO3- secretion and stimulate the insertion of aquaporin water channels into the cholangiocyte's apical membrane. Flow of bile is thereby increased during the postprandial period, when bile acids are needed to aid in assimilation of lipid.
|
| Figure 31-9 Pathways for entry of solutes into bile. (Modified from Barrett KE: Gastrointestinal Physiology. New York, McGraw-Hill, 2006.) |
| page 548 | | | page 549 |
| Figure 31-10 The major transport processes of cholangiocytes that secrete an alkaline-rich fluid and reclaim useful substances. |
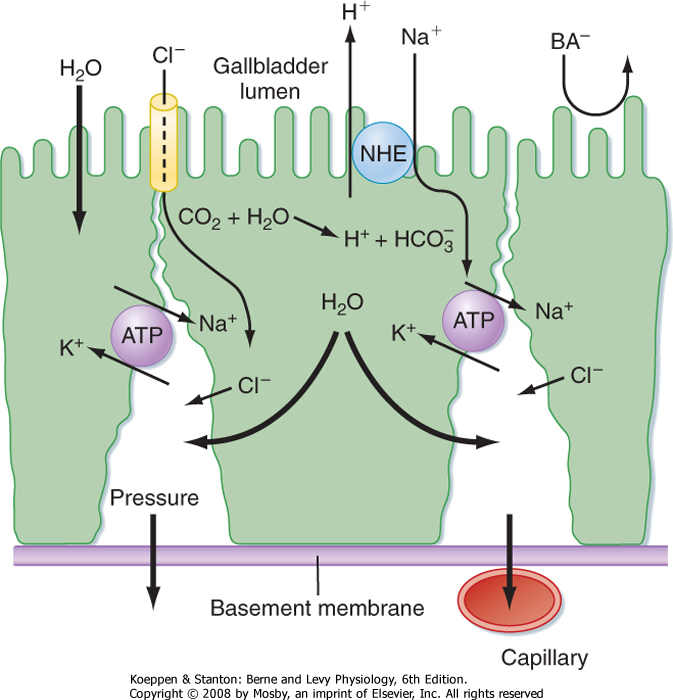
| Figure 31-11 Mechanisms accounting for concentration of bile during storage in the gallbladder. |
| Humans are unusually susceptible to gallstones, which represent precipitated bile constituents that accumulate in the gallbladder or elsewhere in the biliary tree. Gallstones are composed predominantly of cholesterol or Ca++ bilirubinate (cholesterol versus pigment stones, respectively). Their significance lies in their propensity to obstruct biliary flow and thereby result in pain, poor tolerance of large fatty meals, retention of biliary constituents, and (if left untreated) liver injury. In susceptible individuals, mechanisms that normally prevent nucleation of saturated bile are either defective or overcome, and small crystals form and can grow into gallstones. Human bile is often supersaturated in terms of its cholesterol content, thus increasing the risk for stone formation, particularly during prolonged fasting. Gallstones are especially common in middle-aged women who are obese, particularly those who have borne children, for unknown reasons. In severe cases, the gallbladder may be removed surgically, which is usually accomplished laparoscopically. Small gallstones that have lodged in the biliary tree can sometimes be retrieved endoscopically by inserting a small snare through the sphincter of Oddi from an endoscope. |
|
| Finally, bile enters the ducts and is conveyed toward the intestine. However, in the period between meals, outflow is blocked by constriction of the sphincter of Oddi, and thus bile is redirected to the gallbladder. The gallbladder is a muscular sac lined with high-resistance epithelial cells. During gallbladder storage, bile becomes concentrated because sodium ions are
actively absorbed in exchange for protons, and bile acids, as the major anions, are too large to exit across the gallbladder epithelial tight junctions (Fig. 31-11). However, although the concentration of bile acids can rise more than 10-fold, bile remains isotonic because a single micelle acts as only one osmotically active particle. Any additional bile acid monomers that become available as a result of concentration are thus immediately incorporated into existing mixed micelles. This also reduces, to some extent, the risk that cholesterol
will precipitate from bile. However, cholesterol is supersaturated in the bile of many adults, with precipitation normally being inhibited by the presence of antinucleating proteins. Prolonged storage of bile increases the chance that nucleation can occur, thus making a good case for never skipping breakfast and perhaps explaining why gallstone disease is relatively prevalent in humans.
|
| page 549 | | | page 550 |
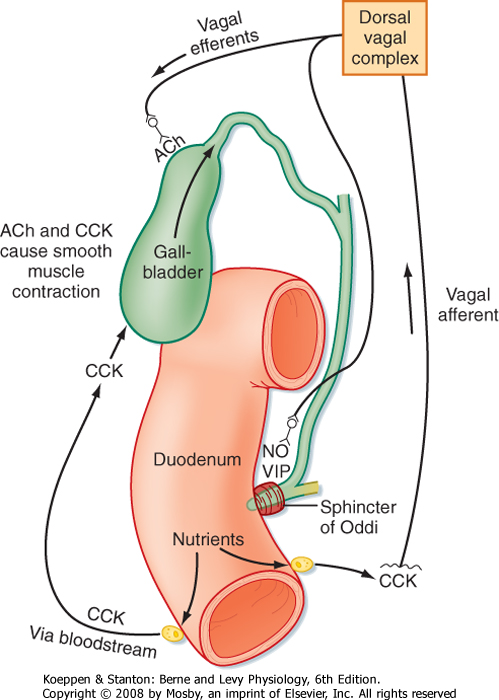
| Figure 31-12 Neurohumoral control of gallbladder contraction and biliary secretion. The pathway also involves relaxation of the sphincter of Oddi to permit outflow of bile into the duodenum. ACh, acetylcholine; CCK, cholecystokinin; NO, nitric oxide; VIP, vasoactive intestinal polypeptide. |
| Bile is secreted from the gallbladder in response to signals that simultaneously relax the sphincter of Oddi
and contract the smooth muscle that encircles the gallbladder epithelium (Fig. 31-12). A critical mediator of this response is cholecystokinin-indeed, this hormone was named for its ability to contract the gallbladder. In addition, intrinsic neural reflexes and vagal pathways, some of which themselves are stimulated by the ability of cholecystokinin to bind to vagal afferents, probably also contribute to gallbladder contractility. The net result is ejection of a concentrated bolus of bile into the duodenal lumen, where the constituent mixed micelles can aid in lipid uptake. Then, when no longer needed, the bile acids are reclaimed and reenter the enterohepatic circulation to begin the cycle again. However, the other components of bile are largely lost in stool, thus providing for their excretion from the body.
|
| Bilirubin Formation and Excretion by the Liver
|
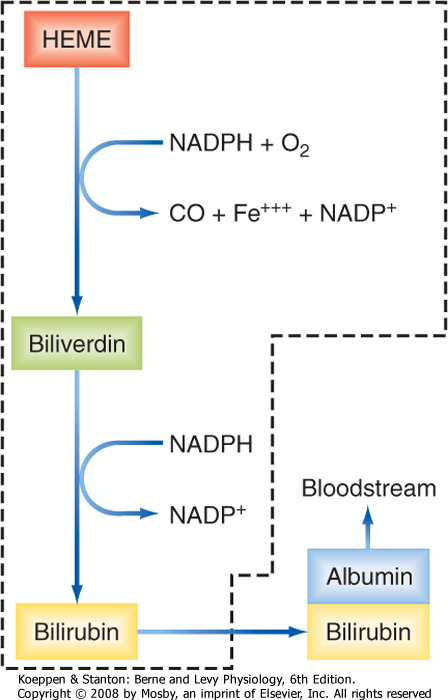
| Figure 31-13 Conversion of heme to bilirubin. The reactions inside the dashed box occur in cells of the reticuloendothelial system. |
| The liver is also important for excretion of bilirubin, which is a metabolite of heme that is potentially toxic to the body. Bilirubin has recently been shown to act as an antioxidant, but it also serves as a way to eliminate the excess heme released from the hemoglobin of senescent red blood cells. Indeed, red blood cells account for 80% of bilirubin production, with the remainder coming from additional heme-containing
proteins in other tissues such as skeletal muscle and the liver itself. Bilirubin is capable of crossing the blood-brain barrier and, if present in excessive levels, results in brain dysfunction for reasons that are not well understood; it can be fatal if left untreated. Bilirubin and its metabolites are also notable for the fact that they provide color to bile, feces, and to a lesser extent, urine. By the same token, when bilirubin accumulates in the circulation as a result of liver disease, it is responsible for the common symptom of jaundice, or yellowing of the skin and conjunctiva.
|
| page 550 | | | page 551 |
| Bilirubin is synthesized from heme by a two-stage reaction that takes place in phagocytic cells of the reticuloendothelial system, including Kupffer cells and cells in the spleen (Fig. 31-13). The enzyme heme oxygenase that is present in these cells liberates iron from the heme molecule and produces the green pigment biliverdin. This, in turn, can be reduced to form yellow bilirubin. Because this molecule is essentially insoluble in aqueous solutions at neutral pH, it is transported through the bloodstream bound to albumin. When this complex reaches the liver, it enters the space of Disse, where bilirubin is selectively taken up across the basolateral membrane of hepatocytes via an OATP transporter (Table 31-1). In the microsomal compartment, bilirubin is then conjugated with one or two molecules of glucuronic acid to enhance its aqueous solubility. The reaction is catalyzed by UDP glucuronyl transferase (UGT). This enzyme is synthesized only slowly after birth, which explains why mild jaundice is relatively common in newborn infants. The bilirubin conjugates are then secreted into bile by a multidrug-related protein (MRP2) located in the canalicular membrane. Notably, the conjugated forms of bilirubin cannot be reabsorbed from the intestine, thereby ensuring that they can be excreted.
However, transport of bilirubin across the hepatocyte, and indeed its initial uptake from the bloodstream, is relatively inefficient, so some conjugated and unconjugated bilirubin is present in plasma even under normal conditions. Both circulate bound to albumin, but the conjugated form is bound more loosely and thus can enter the urine.
|
| In the colon, bilirubin conjugates are deconjugated by bacterial enzymes, whereupon the bilirubin liberated is metabolized by bacteria to yield urobilinogen, which is reabsorbed, and urobilins and stercobilins, which are excreted. Absorbed urobilinogen, in turn, can be taken up by hepatocytes and reconjugated, thus giving the molecule yet another chance to be excreted.
|
| Crigler-Najjar syndrome is a condition associated with mutations in the hepatocyte enzyme UGT. In type I Crigler-Najjar syndrome, a congenital missense mutation results in complete lack of this enzyme, whereas patients with type II Crigler-Najjar syndrome have a milder mutation that reduces UGT levels to about 10% of those seen in normal individuals. Thus, with varying degrees of severity, Crigler-Najjar syndrome impairs the ability of hepatocytes to conjugate bilirubin. The unconjugated bilirubin regurgitates back into the circulation and binds to albumin, with an associated risk of neurologic injury if levels rise precipitously. The only effective treatment of type I Crigler-Najjar syndrome at present is liver transplantation; those with type II disease can sometimes be managed effectively with blue light. This converts circulating unconjugated bilirubin to forms that are more water soluble and thus less firmly bound to albumin, which can be excreted in urine. |
|
| Measurement of bilirubin in plasma, as well as assessment of whether it is unconjugated or conjugated, is an important tool in the evaluation of liver
disease. The presence of unconjugated bilirubin, which is essentially fully albumin bound and cannot be excreted in urine, reflects either loss of UGT (or a normal, temporary delay in its maturation in infants) or a sudden oversupply of heme that overwhelms the conjugation mechanism (such as occurs in transfusion reactions or in Rhesus-incompatible newborns). Conjugated bilirubinemia, on the other hand, is characterized by the presence of bilirubin in urine, to which it imparts a dark coloration. This is indicative of genetic defects in the transporter that mediates bilirubin glucuronide/diglucuronide secretion into the canaliculus, or it may be due to blockage to flow of bile, perhaps caused by an obstructing gallstone. In both cases, bilirubin conjugates are formed in the liver, but with no means of exit, they regurgitate back into plasma for urinary excretion.
|
| AMMONIA HANDLING BY THE LIVER
|
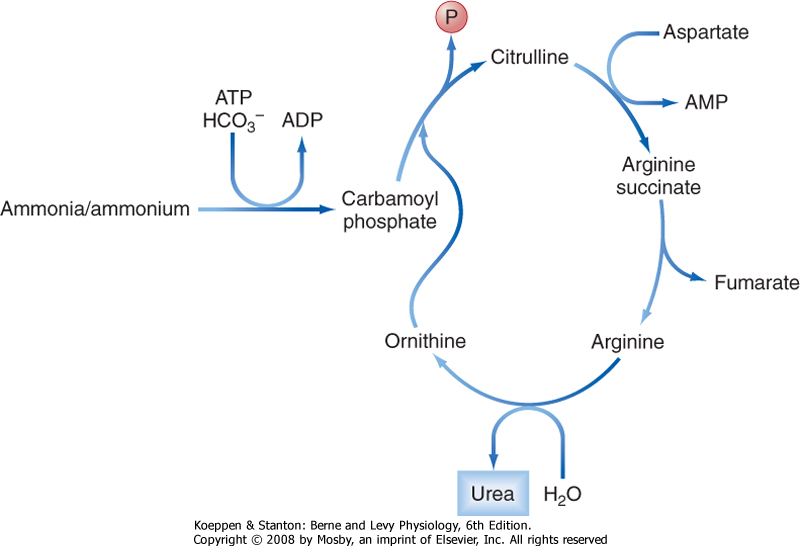
| Ammonia (NH3) is a small, neutral metabolite that arises from protein catabolism and bacterial activity and is highly membrane permeant. The liver is a critical contributor to the prevention of ammonia accumulation in the circulation, which is important because like bilirubin, ammonia is toxic to the central nervous system. The liver eliminates ammonia from the body by converting it to urea via a series of enzymatic reactions known as the urea, or Krebs-Henseleit, cycle (Fig. 31-14). The liver is the only tissue in the body that can convert ammonia to urea.
|
| Figure 31-14 The urea cycle. |
| page 551 | | | page 552 |
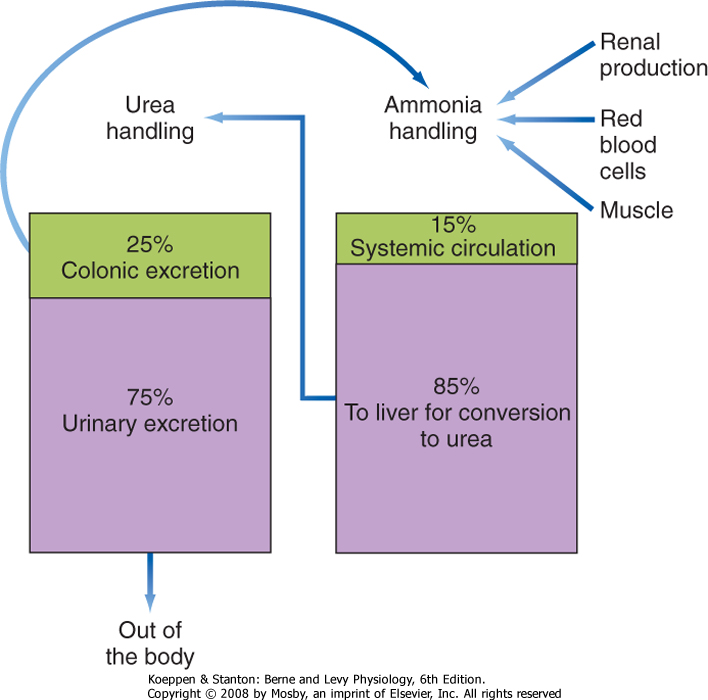
| Figure 31-15 Ammonia homeostasis in health. |
| Ammonia is derived from two major sources. Approximately 50% is produced in the colon by bacterial ureases. Because the colonic lumen is normally slightly acidic, some of this ammonia is converted to the ammonium ion (NH4+), which renders it impermeant to the colonic epithelium and therefore allows it to be excreted in stool. However, the remainder of the ammonia generated crosses the colonic epithelium passively and is transported to the liver via the portal
circulation. The other major source of ammonia (approximately 40%) is the kidney (see Chapter 36). A small amount of ammonia (approximately 10%) is derived from deamination of amino acids in the liver itself, by metabolic processes in muscle cells, and via the release of glutamine from senescent red blood cells.
|
| The "mass balance" for ammonia handling in a healthy adult is presented in Figure 31-15. As just noted, ammonia is a small, neutral molecule that readily crosses cell membranes without the benefit of a specific transporter, although some membrane proteins transport ammonia, including certain aquaporins. Whatever the mechanism for transport, the physicochemical properties of ammonia ensure that it is efficiently extracted from the portal and systemic circulation by hepatocytes, where it then enters the urea cycle to be converted to urea (Fig. 31-14) and is subsequently transported back into the systemic circulation. Urea is a small, neutral molecule that is readily filtered at the glomerulus, and it is reabsorbed by the kidney tubules such that approximately 50% of the filtered urea is excreted in urine (see Chapter 36). Urea that enters the colon is either excreted or metabolized to ammonia via colonic bacteria, with the resulting ammonia being reabsorbed or excreted.
|
| If the metabolic capacity of the liver is compromised acutely, coma and death can rapidly ensue. In chronic liver disease, patients may experience a gradual decline in mental function that reflects the action of both ammonia and other toxins that cannot be cleared by the liver, in a condition known as hepatic encephalopathy. The development of confusion, dementia, and eventually coma in a patient with liver disease is evidence of significant progression, and these symptoms can prove fatal if left untreated.
|
| CLINICAL ASSESSMENT OF LIVER FUNCTION
|
| Given the importance of the liver for homeostasis, tests of liver function are a mainstay of clinical diagnosis. Such tests have several goals: (1) to assess whether hepatocytes have been injured or are dysfunctional, (2) to determine whether bile excretion has been interrupted, and (3) to evaluate whether cholangiocytes have been injured or are dysfunctional. Liver function tests are also used to monitor responses to therapy or rejection reactions after liver transplantation. However, not all such tests measure function directly. Nevertheless, liver function tests are discussed briefly because of their link to hepatic physiology.
|
| Tests for hepatocyte injury rely on markers that are specific for this cell type. When hepatocytes are killed by necrotic responses to inflammation or infection, for example, they release enzymes, including alanine aminotransferase (ALT) and aspartate aminotransferase (AST). These enzymes, which are essential to interconvert amino acids, are easily measured in serum and indicate hepatocyte injury, although AST may also be released after injury to other tissues, including the heart. Two other tests are markers of injury to the biliary system. Alkaline phosphatase is expressed in the canalicular membrane, and elevations of this enzyme in plasma suggest localized obstruction to bile flow. Similarly, increased levels of GGT are seen when there is damage to cholangiocytes.
|
| Measurement of bilirubin in the circulation or in urine also provides an insight into liver function. In addition, measurement of any of the other characteristic secreted products of the liver can be used to diagnose liver disease. Clinically, the most common tests are measurements of serum albumin and a blood clotting parameter, the prothrombin time. If results of these tests are abnormal, when considered together with other aspects of the clinical picture, a diagnosis of liver disease may be established. Blood glucose and ammonia levels are frequently monitored in patients with chronic liver disease. Finally, imaging tests and histological examination of biopsy specimens of liver parenchyma, usually obtained percutaneously, are also important in the evaluation and monitoring of patients with suspected or proven liver disease.
|
| page 552 | | | page 553 |
- Vital functions of the liver include carbohydrate, lipid, and protein metabolism and synthesis; detoxification of unwanted substances; and excretion of circulating substances that are lipid soluble and carried in the bloodstream bound to albumin. The liver also synthesizes the majority of plasma proteins, including albumin.
- Liver function depends on its unique anatomy, its constituent cell types (especially hepatocytes), and the unusual arrangement of its blood supply.
- Substances are excreted from the liver in bile. Bile flow is driven by the presence of bile acids, which are amphipathic end products of cholesterol metabolism that are produced by hepatocytes. Bile acids circulate between the liver and intestine to conserve their mass, and water-insoluble metabolites, such as cholesterol, are carried in bile in the form of mixed micelles.
- Bile is stored in the gallbladder between meals, where it is concentrated and released when hormonal and neural signals simultaneously contract the gallbladder and relax the sphincter of Oddi.
- The liver is critical for disposing of certain substances that would be toxic if allowed to accumulate in the bloodstream, including bilirubin and ammonia.
|
|
| page 553 | | | page 554 |
|