| 34 Control of Body Fluid Osmolality and Volume
|
| The kidneys maintain the osmolality and volume of body fluids within a narrow range by regulating the excretion of water and NaCl, respectively. This chapter discusses the regulation of renal water excretion (urine concentration and dilution) and NaCl excretion. The composition and volumes of the various body fluid compartments are reviewed in Chapter 2.
|
| CONTROL OF BODY FLUID OSMOLALITY: URINE CONCENTRATION AND DILUTION
|
| As described in Chapter 2, water constitutes approximately 60% of the healthy adult human body. Body water is divided into two compartments (i.e., intracellular fluid [ICF] and extracellular fluid [ECF]), which are in osmotic equilibrium. Water intake into the body generally occurs orally. However, in clinical situations, intravenous infusion is an important route of water entry.
|
| The kidneys are responsible for regulating water balance and under most conditions are the major route for elimination of water from the body (Table 34-1). Other routes of water loss from the body include evaporation from cells of the skin and respiratory passages. Collectively, water loss by these routes is termed insensible water loss because the individual is unaware of its occurrence. The production of sweat accounts for the loss of additional water. Water loss by this mechanism can increase dramatically in a hot environment, with exercise, or in the presence of fever (Table 34-2). Finally, water can be lost from the gastrointestinal tract. Fecal water loss is normally small (≈100 mL/day) but can increase dramatically with diarrhea (e.g., 20 L/day with cholera). Vomiting can also cause gastrointestinal water loss.
|
| Although water loss from sweating, defecation, and evaporation from the lungs and skin can vary with environmental conditions or during pathological conditions, loss of water by these routes cannot be regulated. In contrast, the renal excretion of water is tightly regulated to maintain whole-body water balance. Maintenance of water balance requires that water intake and loss from the body be precisely matched. If intake exceeds loss, positive water balance exists. Conversely, if intake is less than loss, negative water balance exists.
|
| When water intake is low or water loss increases, the kidneys conserve water by producing a small volume of urine that is hyperosmotic with respect to plasma. When water intake is high, a large volume of hypoosmotic urine is produced. In a normal individual, urine osmolality (Uosm) can vary from approximately 50 to 1200 mOsm/kg H2O, and the corresponding urine volume can vary from approximately 18 to 0.5 L/day.
|
| It is important to recognize that disorders in water balance are manifested by alterations in body fluid osmolality, which are usually measured by changes in plasma osmolality (Posm). Because the major determinant of plasma osmolality is Na+ (with its anions Cl- and HCO3-), these disorders also result in alterations in plasma [Na+]. When an abnormal plasma [Na+] is observed in an individual, it is tempting to suspect a problem in Na+ balance. However, the problem most often relates to water balance, not Na+ balance. As described later, changes in Na+ balance result in alterations in the volume of ECF, not its osmolality.
|
| Under steady-state conditions, the kidneys control water excretion independently of their ability to control the excretion of various other physiologically important substances such as Na+, K+, and urea. Indeed, this ability is necessary for survival because it allows water balance to be achieved without upsetting the other homeostatic functions of the kidneys.
|
| The following sections discuss the mechanisms by which the kidneys excrete either hypoosmotic (dilute) or hyperosmotic (concentrated) urine. The control of vasopressin secretion and its important role in regulating excretion of water by the kidneys are also explained (see also Chapter 40).
|
| Antidiuretic hormone (ADH), or vasopressin, acts on the kidneys to regulate the volume and osmolality of urine. When plasma ADH levels are low, a large volume of urine is excreted (diuresis), and the urine is dilute.* When plasma levels are high, a small volume of urine is excreted (antidiuresis), and the urine is concentrated.
|
| page 594 |  | | page 595 |
|
Table 34-1.
Normal Routes of Water Gain and Loss in Adults at Room Temperature (23° C) |
| Route | mL/Day |
| Water Intake |
| Fluid* | 1200 |
| In food | 1000 |
| Metabolically produced from food | 300 |
| TOTAL | 2500 |
| Water Output |
| Insensible | 700 |
| Sweat | 100 |
| Feces | 200 |
| Urine | 1500 |
| TOTAL | 2500 |
*Fluid intake varies widely for both social and cultural reasons.
|
| In the clinical setting, hypoosmolality (a reduction in plasma osmolality) shifts water into cells, and this process results in cell swelling. Symptoms associated with hypoosmolality are related primarily to swelling of brain cells. For example, a rapid fall in Posm can alter neurological function and thereby cause nausea, malaise, headache, confusion, lethargy, seizures, and coma. When Posm is increased (i.e., hyperosmolality), water is lost from cells. The symptoms of an increase in Posm are also primarily neurological and include lethargy, weakness, seizures, coma, and even death. |
| Symptoms associated with changes in body fluid osmolality vary depending on how quickly the osmolality is changed. Rapid changes in osmolality (i.e., over a period of hours) are less well tolerated than changes that occur more gradually (i.e., over a period of days to weeks). Indeed, individuals in whom alterations in body fluid osmolality have developed over an extended period may be entirely asymptomatic. This reflects the ability of cells over time to either eliminate intracellular osmoles, as occurs with hypoosmolality, or generate new intracellular osmoles in response to hyperosmolality and thus minimize changes in cell volume of the neurons. This has important clinical implications when treating a patient with abnormal plasma osmolality. For example, rapid correction of osmolality of an individual who has had long-standing hypoosmolality of body fluids can lead to demyelination, especially of the pons, the results of which are irreversible. Depending on the extent of pontine demyelination, this condition can be fatal. |
|
|
Table 34-2.
Effect of Environmental Temperature and Exercise on Water Loss and Intake (mL/day) in Adults |
| | Normal Temperature | Hot Weather* | Prolonged Heavy Exercise* |
| Water Loss |
| Insensible Loss |
| Skin | 350 | 350 | 350 |
| Lungs | 350 | 250 | 650 |
| Sweat | 100 | 1400 | 5000 |
| Feces | 200 | 200 | 200 |
| Urine* | 1500 | 1200 | 500 |
| Total Loss | 2500 | 3400 | 6700 |
| Water Intake to Maintain Water Balance | 2500 | 3400 | 6700 |
*In hot weather and during prolonged heavy exercise, water balance is maintained by increased water ingestion. Decreased excretion of water by the kidneys alone is insufficient to maintain water balance.
|
| The gene for ADH is found on chromosome 20. It contains approximately 2000 base pairs with three exons and two introns. The gene codes for a preprohormone that consists of a signal peptide, the ADH molecule, neurophysin, and a glycopeptide (copeptin). As the cell processes the preprohormone, the signal peptide is cleaved off in the rough endoplasmic reticulum. Once packaged in neurosecretory granules, the preprohormone is further cleaved into ADH, neurophysin, and copeptin molecules. The neurosecretory granules are then transported down the axon to the posterior pituitary and stored in the nerve endings until released. When the neurons are stimulated to secrete ADH, the action potential opens Ca++ channels in the nerve terminal, which raises intracellular [Ca++] and causes exocytosis of the neurosecretory granules. All three peptides are secreted in this process. Neurophysin and copeptin do not have an identified physiological function. |
|
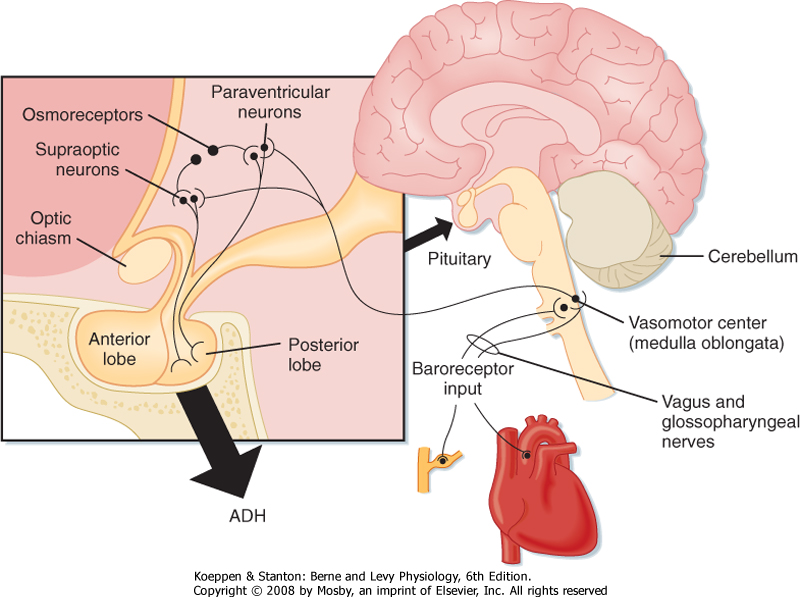
| ADH is a small peptide that is nine amino acids in length. It is synthesized in neuroendocrine cells located within the supraoptic and paraventricular nuclei of the hypothalamus.* The synthesized hormone is packaged in granules that are transported down the
axon of the cell and stored in nerve terminals located in the neurohypophysis (posterior pituitary). The anatomy of the hypothalamus and pituitary gland is shown in Figure 34-1.
|
| Secretion of ADH by the posterior pituitary can be influenced by several factors. The two primary physiological regulators of ADH secretion are the osmolality of the body fluids (osmotic) and the volume and pressure of the vascular system (hemodynamic). Other factors that can alter ADH secretion include nausea (stimulates), atrial natriuretic peptide (inhibits), and angiotensin II (stimulates). A number of drugs, prescription and nonprescription, also affect secretion of ADH. For example, nicotine stimulates secretion, whereas ethanol inhibits secretion.
|
| page 595 | | | page 596 |
| Figure 34-1 Anatomy of the hypothalamus and pituitary gland (midsagittal section). Also shown are pathways involved in regulating secretion of ADH. Afferent fibers from the baroreceptors are carried in the vagus and glossopharyngeal nerves. The closed box is an expanded view of the hypothalamus and pituitary gland. |
| Osmotic Control of ADH Secretion
|
| Changes in the osmolality of body fluids play the most important role in regulating secretion of ADH; changes as minor as 1% are sufficient to alter it significantly. Although neurons in the supraoptic and paraventricular nuclei respond to changes in body fluid osmolality by altering their secretion of ADH, it is clear that there are separate cells in the anterior hypothalamus that are exquisitely sensitive to changes in body fluid osmolality and therefore play an important role in regulating the secretion of ADH.* These cells, termed osmoreceptors, appear to behave as osmometers and sense changes in body fluid osmolality by either shrinking or swelling. The osmoreceptors respond only to solutes in plasma that are effective osmoles (see Chapter 1). For example, urea is an ineffective osmole when the function of osmoreceptors is considered. Thus, elevation of the plasma urea concentration alone has little effect on ADH secretion.
|
| When the effective osmolality of plasma increases, the osmoreceptors send signals to ADH-synthesizing/secreting cells located in the supraoptic and paraventricular nuclei of the hypothalamus, and synthesis and secretion of ADH are stimulated. Conversely, when the effective osmolality of plasma is reduced, secretion is inhibited. Because ADH is rapidly degraded in plasma, circulating levels can be reduced to zero within minutes after secretion is inhibited. As a result, the ADH system can respond rapidly to fluctuations in body fluid osmolality.
|
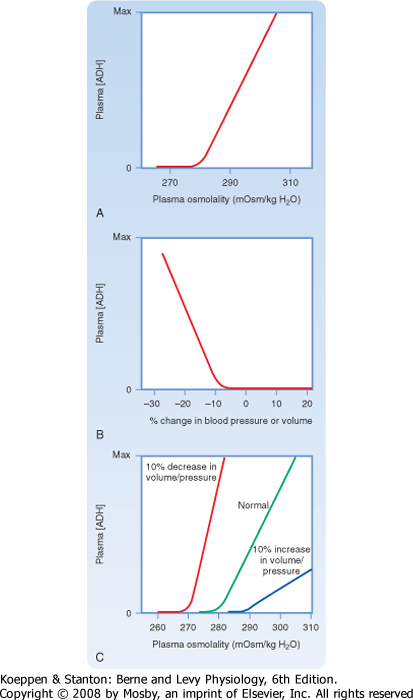
| Figure 34-2, A, illustrates the effect of changes in plasma osmolality on circulating ADH levels. The slope of the relationship is quite steep and accounts for the sensitivity of this system. The set point of the system is the plasma osmolality value at which ADH secretion begins to increase. Below this set point, virtually no ADH is released. The set point varies among individuals and is genetically determined. In healthy adults, it varies from 275 to 290 mOsm/kg H2O (average, ≈280 to 285 mOsm/kg H2O). Several physiological factors can also change the set point in a given individual. As discussed later, alterations in blood volume and pressure can shift it. In addition, pregnancy is associated with a decrease in the set point.
|
| Hemodynamic Control of ADH Secretion
|
| page 596 | | | page 597 |
| Figure 34-2 Osmotic and hemodynamic control of secretion of ADH. A, Effect of changes in plasma osmolality (constant blood volume and pressure) on plasma ADH levels. B, Effect of changes in blood volume or pressure (constant plasma osmolality) on plasma ADH levels. C, Interactions between osmolality and blood volume and pressure stimuli on plasma ADH levels. |
| A decrease in blood volume or pressure also stimulates secretion of ADH. The receptors responsible for this response are located in both the low-pressure (left atrium and large pulmonary vessels) and the high-pressure (aortic arch and carotid sinus) sides of the circulatory system. Because the low-pressure receptors are located in the high-compliance side of the circulatory system (i.e., venous) and because the majority of blood is in the venous side of the circulatory system, these low-pressure receptors can be viewed as responding to overall vascular volume. The high-pressure receptors respond to arterial pressure. Both groups of receptors are sensitive to stretch of the wall of the structure in which they are located (e.g., cardiac atrial wall, wall of the aortic arch) and are termed baroreceptors. Signals from these receptors are carried in afferent fibers of the vagus and glossopharyngeal nerves to the brainstem (solitary tract nucleus of the medulla oblongata), which is part of the center that regulates heart rate and blood pressure (see also Chapter 18). Signals are then relayed from the brainstem to the ADH-secreting cells of the supraoptic and paraventricular hypothalamic nuclei. The
sensitivity of the baroreceptor system is less than that of the osmoreceptors, and a 5% to 10% decrease in blood volume or pressure is required before ADH secretion is stimulated. This is illustrated in Figure 34-2, B. A number of substances have been shown to alter the secretion of ADH through their effects on blood pressure, including bradykinin and histamine, which lower pressure and thus stimulate ADH secretion, and norepinephrine, which increases blood pressure and inhibits ADH secretion.
|
| Alterations in blood volume and pressure also affect the response to changes in body fluid osmolality (see Fig. 34-2, C). With a decrease in blood volume or pressure, the set point is shifted to lower osmolality values and the slope of the relationship is steeper. In terms of survival of the individual, this means that when faced with circulatory collapse, the kidneys will continue to conserve water, even though by doing so they reduce the osmolality of body fluids. With an increase in blood volume or pressure, the opposite occurs. The set point is shifted to higher osmolality values, and the slope is decreased.
|
| Actions of ADH on the Kidneys
|
| The primary action of ADH on the kidneys is to increase the permeability of the collecting duct to water. In addition and importantly, ADH increases the permeability of the medullary portion of the collecting duct to urea. Finally, ADH stimulates reabsorption of NaCl by the thick ascending limb of Henle's loop, the distal tubule, and the collecting duct.
|
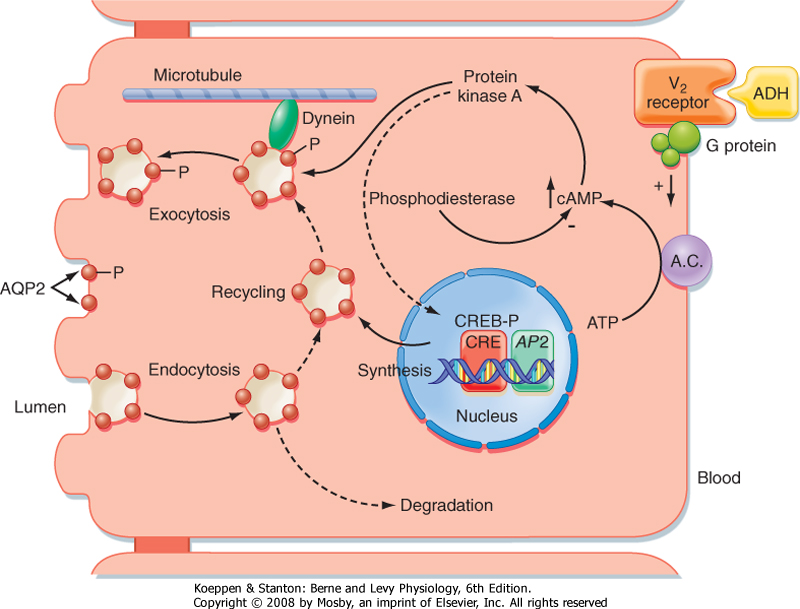
| The actions of ADH on permeability of the collecting duct to water have been studied extensively. ADH binds to a receptor on the basolateral membrane of the cell. This receptor is termed the V2 receptor (i.e., vasopressin 2 receptor).* Binding to this receptor, which is coupled to adenylyl cyclase via a stimulatory G protein (Gs), increases intracellular levels of cAMP. The rise in intracellular cAMP activates protein kinase A (PKA), which ultimately results in the insertion of vesicles containing aquaporin-2 (AQP2) water channels into the apical membrane of the cell, as well as the synthesis of more AQP2 (Fig. 34-3). With the removal of ADH, these water channels are reinternalized into the cell, and the apical membrane is once again impermeable to water. This shuttling of water channels into and out of the apical membrane provides a rapid mechanism for controlling permeability of the membrane to water. Because the basolateral membrane is freely permeable to water as a result of the presence of AQP3 and AQP4 water channels, any water that enters the cell through apical membrane water channels exits across the basolateral membrane, thereby resulting in net absorption of water from the tubule lumen.
|
| page 597 | | | page 598 |
| Inadequate release of ADH from the posterior pituitary results in the excretion of large volumes of dilute urine (polyuria). To compensate for this loss of water, the individual must ingest large volumes of water (polydipsia) to maintain constant body fluid osmolality. If the individual is deprived of water, the body fluids will become hyperosmotic. This condition is called central diabetes insipidus or pituitary diabetes insipidus. Central diabetes insipidus can be inherited, although this is rare. It occurs more commonly after head trauma and with brain neoplasms or infections. Individuals with central diabetes insipidus have a urine-concentrating defect that can be corrected by the administration of exogenous ADH. |
| The inherited (autosomal dominant) form of central diabetes insipidus has been shown to represent multiple mutations in the ADH gene. In patients with this form of central diabetes insipidus, mutations have been identified in all regions of the ADH gene (i.e., ADH, copeptin, and neurophysin). The most common mutation is found in the neurophysin portion of the gene. In each of these situations there is defective trafficking of the peptide, with abnormal accumulation in the endoplasmic reticulum. It is believed that this abnormal accumulation in the endoplasmic reticulum results in death of the ADH secretory cells of the supraoptic and paraventricular nuclei. |
| The syndrome of inappropriate ADH secretion (SIADH) is a common clinical problem characterized by plasma ADH levels that are elevated above what would be expected on the basis of body fluid osmolality and blood volume and pressure-hence the term inappropriate ADH secretion. Individuals with SIADH retain water, and their body fluids become progressively hypoosmotic. In addition, their urine is more hyperosmotic than expected based on the low body fluid osmolality. SIADH can be caused by infections and neoplasms of the brain, drugs (e.g., antitumor drugs), pulmonary diseases, and carcinoma of the lung. Many of these conditions stimulate secretion of ADH by altering neural input to the ADH secretory cells. However, small cell carcinoma of the lung produces and secretes a number of peptides, including ADH. |
|
| In addition to the acute effects of ADH just described, ADH regulates the expression of AQP2 (and AQP3). When large volumes of water are ingested over an
extended period (e.g., psychogenic polydipsia), expression of AQP2 and AQP3 in the collecting duct is reduced. As a consequence, when water ingestion is restricted, these individuals cannot maximally concentrate their urine. Conversely, in states of restricted water ingestion, expression of AQP2 and AQP3 in the collecting duct increases and thus facilitates the excretion of maximally concentrated urine.
|
| The gene for the V2 receptor is located on the X chromosome. It codes for a 371-amino acid protein that is in the family of receptors that have seven membrane-spanning domains and are coupled to heterotrimeric G proteins. As shown in Figure 34-3, binding of ADH to its receptor on the basolateral membrane activates adenylyl cyclase. The increase in intracellular cAMP then activates protein kinase (PKA), which results in phosphorylation of AQP2 water channels, as well as increased transcription of the AQP2 gene via activation of a cAMP-response element (CRE). Vesicles containing phosphorylated AQP2 move toward the apical membrane along microtubules driven by the molecular motor dynein. Once near the apical membrane, proteins called SNAREs interact with vesicles containing AQP2 and facilitate fusion of these vesicles with the membrane. The addition of AQP2 to the membrane allows water to enter the cell driven by the osmotic gradient (lumen osmolality < cell osmolality). The water then exits the cell across the basolateral membrane through AQP3 and AQP4 water channels, which are constitutively present in the basolateral membrane. When the V2 receptor is not occupied by ADH, the AQP2 water channels are removed from the apical membrane by clathrin-mediated endocytosis, thus rendering the apical membrane once again impermeable to water. The endocytosed AQP2 molecules may be either stored in cytoplasmic vesicles, ready for reinsertion into the apical membrane when ADH levels in plasma increase, or degraded. |
| Recently, individuals have been found who have activating (gain-of-function) mutations in the V2 receptor gene. Thus, the receptor is constitutively activated, even in the absence of ADH. These individuals have laboratory findings similar to those seen in SIADH, including reduced plasma osmolality, hyponatremia (reduced plasma [Na+]), and urine more concentrated than would be expected from the reduced body fluid osmolality. However, unlike SIADH, where circulating levels of ADH are elevated and thus responsible for water retention by the kidneys, these individuals have undetectable levels of ADH in their plasma. This new clinical entity has been termed "nephrogenic syndrome of inappropriate antidiuresis." |
|
| It is also clear that expression of AQP2 (and in some instances also AQP3) varies in pathological conditions associated with disturbances in urine concentration
and dilution. As discussed elsewhere, AQP2 expression is reduced in a number of conditions associated with impaired urine-concentrating ability. By contrast, in conditions associated with water retention, such as congestive heart failure, hepatic cirrhosis, and pregnancy, AQP2 expression is increased.
|
| page 598 | | | page 599 |
| Figure 34-3 Action of ADH via the V2 receptor on the principal cell of the late distal tubule and collecting duct. See text for details. A.C., adenylyl cyclase; AP2, aquaporin-2 gene; AQP2, aquaporin-2; CRE, cAMP response element; CREB-P, phosphorylated cAMP response element-binding protein; -P, phosphorylated proteins. (Adapted and modified from Brown D, Nielsen S. In Brenner BM [ed]: The Kidney, 7th ed. Philadelphia, Saunders, 2004.) |
| ADH also increases the permeability of the terminal portion of the inner medullary collecting duct to urea. This results in an increase in reabsorption of urea and an increase in the osmolality of the medullary interstitial fluid. The apical membrane of medullary collecting
duct cells contains two different urea transporters (UT-A1 and UT-A3).* ADH, acting through the cAMP/PKA cascade, increases permeability of the apical membrane to urea. This increase in permeability is associated with phosphorylation of UT-A1 and perhaps also UT-A3. Increasing the osmolality of the interstitial fluid of the renal medulla also increases the permeability of the collecting duct to urea. This effect is mediated by the phospholipase C pathway and involves phosphorylation of protein kinase C. Thus, this effect is separate and additive to that of ADH.
|
| In addition to its acute effect on permeability of the collecting duct to urea, ADH also increases the abundance of UT-A1 in states of chronic water restriction. In contrast, with water loading (i.e., suppressed ADH levels), UT-A1 abundance in the collecting duct is reduced.
|
| ADH also stimulates reabsorption of NaCl by the thick ascending limb of Henle's loop and by the distal tubule and cortical segment of the collecting duct. This increase in Na+ reabsorption is associated with increased abundance of key Na+ transporters: 1Na+-1K+-2Cl- symporter (thick ascending limb of Henle's loop), Na+-Cl- symporter (distal tubule), and the epithelial Na+ channel (ENaC, in the distal tubule and collecting duct). It is thought that stimulation of NaCl transport by the thick ascending limb may help maintain the hyperosmotic medullary interstitium that is necessary for the absorption of water from the medullary portion of the collecting duct (see later).
|
| In addition to affecting the secretion of ADH, changes in plasma osmolality and blood volume or pressure lead to alterations in the perception of thirst. When body fluid osmolality is increased or blood volume or pressure is reduced, the individual perceives thirst. Of these stimuli, hypertonicity is the more potent. An increase in plasma osmolality of only 2% to 3% produces a strong desire to drink, whereas decreases in blood volume and pressure in the range of 10% to 15% are required to produce the same response.
|
| As already discussed, there is a genetically determined threshold for ADH secretion (i.e., a body fluid osmolality above which ADH secretion increases). Similarly, there is a genetically determined threshold for triggering the sensation of thirst. However, the thirst threshold is higher than the threshold for ADH secretion. On average, the threshold for ADH secretion is approximately 285 mOsm/kg H2O, whereas the thirst threshold is approximately 295 mOsm/kg H2O. Because of this difference, thirst is stimulated at a body fluid osmolality at which secretion of ADH is already maximal.
|
| page 599 | | | page 600 |
| The neural centers involved in regulating water intake (the thirst center) are located in the same region of the hypothalamus involved in regulating ADH secretion. However, it is not certain whether the same cells serve both functions. Indeed, the thirst response, like the regulation of ADH secretion, occurs only in response to effective osmoles (e.g., NaCl). Even less is known about the pathways involved in the thirst response to decreased blood volume or pressure, but it is believed that they are the same as those involved in the volume- and pressure-related regulation of ADH secretion. Angiotensin II, acting on cells of the thirst
center (subfornical organ), also evokes the sensation of thirst. Because angiotensin II levels are increased when blood volume and pressure are reduced, this effect of angiotensin II contributes to the homeostatic response that restores and maintains body fluids at their normal volume.
|
| The collecting ducts of some individuals do not respond normally to ADH. These individuals cannot maximally concentrate their urine and consequently have polyuria and polydipsia. This clinical entity is termed nephrogenic diabetes insipidus to distinguish it from central diabetes insipidus. Nephrogenic diabetes insipidus can result from a number of systemic disorders and, more rarely, occurs as a result of inherited disorders. Many of the acquired forms of nephrogenic diabetes insipidus are the result of decreased expression of AQP2 in the collecting duct. Decreased expression of AQP2 has been documented in the urine-concentrating defects associated with hypokalemia, lithium ingestion (some degree of nephrogenic diabetes insipidus develops in 35% of individuals who take lithium for bipolar disorder), ureteral obstruction, a low-protein diet, and hypercalcemia. The inherited forms of nephrogenic diabetes insipidus reflect mutations in the ADH receptor (V2 receptor) or the AQP2 molecule. Of these, approximately 90% of hereditary forms of nephrogenic diabetes insipidus are the result of mutations in the V2 receptor gene, with the other 10% being the result of mutations in the AQP2 gene. Because the gene for the V2 receptor is located on the X chromosome, these inherited forms are X-linked. To date, more than 150 different mutations in the V2 receptor gene have been described. Most of the mutations result in trapping of the receptor in the endoplasmic reticulum of the cell; only a few cases result in the surface expression of a V2 receptor that will not bind ADH. The gene coding for AQP2 is located on chromosome 12 and is inherited as both an autosomal recessive and an autosomal dominant defect. As noted in Chapter 1, aquaporins exist as homotetramers. This homotetramer formation explains the difference between the two forms of nephrogenic diabetes insipidus. In the recessive form, heterozygotes produce both normal AQP2 and defective AQP2 molecules. The defective AQP2 monomer is retained in the endoplasmic reticulum of the cell, and thus the homotetramers that do form contain only normal molecules. Accordingly, mutations in both alleles are required to produce nephrogenic diabetes insipidus. In the autosomal dominant form, the defective monomers can form tetramers with normal monomers, as well as with defective monomers. However, these tetramers are unable to traffic to the apical membrane. |
|
| The sensation of thirst is satisfied by the act of drinking even before sufficient water is absorbed from the gastrointestinal tract to correct the plasma osmolality.
Oropharyngeal and upper gastrointestinal receptors appear to be involved in this response. However, relief of the thirst sensation via these receptors is short lived, and thirst is completely satisfied only when plasma osmolality or blood volume or pressure is corrected.
|
| It should be apparent that the ADH and thirst systems work in concert to maintain water balance. An increase in plasma osmolality evokes drinking and, via ADH action on the kidneys, the conservation of water. Conversely, when plasma osmolality is decreased, thirst is suppressed, and in the absence of ADH, renal water excretion is enhanced. However, most of the time fluid intake is dictated by cultural factors and social situations. This is especially the case when thirst is not stimulated. In this situation, maintenance of normal body fluid osmolality relies solely on the ability of the kidneys to excrete water. How the kidney accomplishes this is discussed in detail in the following sections of this chapter.
|
| With adequate access to water, the thirst mechanism can prevent the development of hyperosmolality. Indeed, it is this mechanism that is responsible for the polydipsia seen in response to the polyuria of both central and nephrogenic diabetes insipidus. |
|
| Water intake is also influenced by social and cultural factors. Thus, individuals will ingest water even in the absence of the thirst sensation. Normally, the kidneys are able to excrete this excess water because they can excrete up to 18 L/day of urine. However, in some instances, the volume of water ingested exceeds the kidneys' capacity to excrete water, especially over short periods. When this occurs, the body fluids become hypoosmotic. An example of how water intake can exceed the capacity of the kidneys to excrete water is long-distance running. A recent study of participants in the Boston Marathon found that hyponatremia developed in 13% of the runners during the course of the race.* This reflected the practice of some runners of ingesting water, or other hypotonic drinks, during the race to remain "well hydrated." In addition, water is produced from the metabolism of glycogen and triglycerides used as fuels by the exercising muscle. Because over the course of the race they ingested, as well as generated through metabolism, more water than their kidneys were able to excrete or was lost by sweating, hyponatremia developed. In some racers the hyponatremia was severe enough to elicit the neurological symptoms described previously. |
|
| page 600 | | | page 601 |
| One can find throughout the popular press the admonition to drink eight 8-oz glasses of water a day (the 8 × 8 recommendation). Drinking this volume of water is said to provide innumerable health benefits. As a result, it seems that everyone now has a water bottle as a constant companion. Although ingesting this volume of water over the course of a day (approximately
2 L) will not harm most individuals, there is no scientific evidence to support the beneficial health claims ascribed to the 8 × 8 recommendation.† Indeed most individuals get adequate amounts of water through the food that they ingest and the fluids taken with those meals. |
The maximum amount of water that can be excreted by the kidneys depends on the amount of solute excreted, which in turn depends on food intake. For example, with maximally dilute urine (Uosm = 50 mOsm/kg H2O), the maximum urine output of 18 L/day will be achieved only if the solute excretion rate is 900 mmol/day.
 |
| If excretion of solute is reduced, as commonly occurs in the elderly with reduced food intake, the maximum urine output will decrease. For example, if solute excretion is only 400 mmol/day, a maximum urine output (at Uosm = 50 mOsm/kg H2O) of only 8 L/day can be achieved. Thus, individuals with reduced food intake have a reduced capacity to excrete water. |
| Renal Mechanisms for Dilution and Concentration of Urine
|
| Under normal circumstances, excretion of water is regulated separately from excretion of solutes. For this to occur, the kidneys must be able to excrete urine that is either hypoosmotic or hyperosmotic with respect to body fluids. This ability to excrete urine of varying osmolality in turn requires that solute be separated from water at some point along the nephron. As discussed in Chapter 33, reabsorption of solute in the proximal tubule results in reabsorption of a proportional amount of water. Hence, solute and water are not separated in this portion of the nephron. Moreover, this proportionality between proximal tubule water and solute reabsorption occurs regardless of whether the kidneys excrete dilute or concentrated urine. Thus, the proximal tubule reabsorbs a large portion of the filtered load of solute and water, but it does not produce dilute or concentrated tubular fluid. The loop of Henle, in particular, the thick ascending limb, is the major site where solute and water are separated. Consequently, excretion of both dilute and concentrated urine requires normal function of the loop of Henle.
|
| Excretion of hypoosmotic urine is relatively easy to understand. The nephron must simply reabsorb solute from the tubular fluid and not allow reabsorption of water to also occur. As just noted and as described in greater detail later, reabsorption of solute without concomitant water reabsorption occurs in the ascending limb of Henle's loop. Under appropriate conditions (i.e., in the absence of ADH), the distal tubule and collecting duct also dilute the tubular fluid.
|
| Excretion of hyperosmotic urine is more complex and thus more difficult to understand. This process in essence involves removing water from the tubular fluid without solute. Because water movement is passive and driven by an osmotic gradient, the kidney must generate a hyperosmotic compartment that then reabsorbs water osmotically from the tubular fluid. The compartment in the kidney that serves this function is the interstitium of the renal medulla. Henle's loop, in particular, the thick ascending limb, is critical for generating the hyperosmotic medullary interstitium. Once established, this hyperosmotic compartment drives reabsorption of water from the collecting duct and thereby concentrates the urine.
|
| Figure 34-4 summarizes the essential features of the mechanisms whereby the kidneys excrete either dilute or concentrated urine. Table 34-3 summarizes the transport and passive permeability properties of the nephron segments involved in these processes.
|
| page 601 | | | page 602 |
| page 602 | | | page 603 |
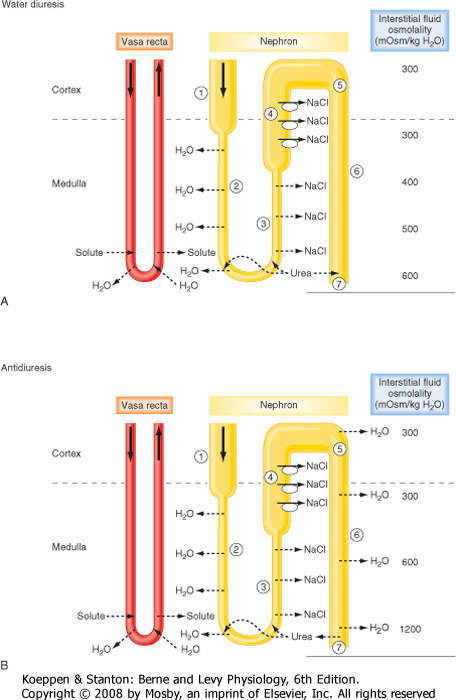
First, how the kidneys excrete dilute urine (water diuresis) when ADH levels are low or zero is considered. The following numbers refer to those encircled in Figure 34-4, A:
- Fluid entering the descending thin limb of the loop of Henle from the proximal tubule is isosmotic with respect to plasma. This reflects the essentially isosmotic nature of solute and water reabsorption in the proximal tubule (see Chapter 33).
- The descending thin limb is highly permeable to water and much less so to solutes such as NaCl and urea. (Note: Urea is an ineffective osmole in many tissues, but it is an effective osmole in many portions of the nephron [Table 34-3]). Consequently, as the fluid in the descending thin limb descends deeper into the hyperosmotic medulla, water is reabsorbed (via AQP1) as a result of the osmotic gradient set up across the descending thin limb by both NaCl and urea, which are present at high concentrations in the medullary interstitium (see later). By this process, tubular fluid at the bend of the loop has an osmolality equal to that of the surrounding interstitial fluid. Although the osmolality of tubular and interstitial fluid is similar at the bend of the loop, their compositions differ. The concentration of NaCl in tubular fluid is greater than that in the surrounding interstitial fluid. However, the concentration of urea in tubular fluid is less than that of interstitial fluid (see later).
- The ascending thin limb is impermeable to water but permeable to NaCl. Consequently, as tubular fluid moves up the ascending limb, NaCl is passively reabsorbed because the concentration of NaCl in tubular fluid is higher than that in interstitial fluid. As a result, the volume of tubular fluid remains unchanged along the length of the thin ascending limb, but the concentration of NaCl
decreases. Thus, as fluid ascends through the thin ascending limb, it becomes less concentrated than the surrounding interstitial fluid (i.e., tubular fluid dilution begins).
- The thick ascending limb of the loop of Henle is impermeable to water and urea. This portion of the nephron actively reabsorbs NaCl from tubular fluid and thereby dilutes it. Dilution occurs to such a degree that this segment is often referred to as the diluting segment of the kidney. Fluid leaving the thick ascending limb is hypoosmotic with respect to plasma (approximately 150 mOsm/kg H2O).
- The distal tubule and the cortical portion of the collecting duct actively reabsorb NaCl and are impermeable to urea. In the absence of ADH, these segments are not permeable to water. Thus, when ADH is absent or present at low levels (i.e., decreased plasma osmolality), the osmolality of tubule fluid in these segments is reduced further because NaCl is reabsorbed without water. Under this condition, fluid leaving the cortical portion of the collecting duct is hypoosmotic with respect to plasma (approximately 50 to 100 mOsm/kg H2O).
- The medullary collecting duct actively reabsorbs NaCl. Even in the absence of ADH, this segment is slightly permeable to water and urea. Consequently, some urea enters the collecting duct from the medullary interstitium, and a small volume of water is reabsorbed.
- Urine has an osmolality as low as approximately 50 mOsm/kg H2O and contains low concentrations of NaCl and urea. The volume of urine excreted can be as much as 18 L/day, or approximately 10% of the glomerular filtration rate (GFR).
|
|
| Figure 34-4 Schematic of the nephron segments involved in dilution and concentration of urine. Henle's loops of juxtamedullary nephrons are shown. A, Mechanism for the excretion of dilute urine (water diuresis). ADH is absent, and the collecting duct is essentially impermeable to water. Note also that during water diuresis the osmolality of the medullary interstitium is reduced as a result of increased vasa recta blood flow and entry of some urea into the medullary collecting duct. B, Mechanism for the excretion of concentrated urine (antidiuresis). Plasma ADH levels are maximal, and the collecting duct is highly permeable to water. Under this condition, the medullary interstitial gradient is maximal. |
|
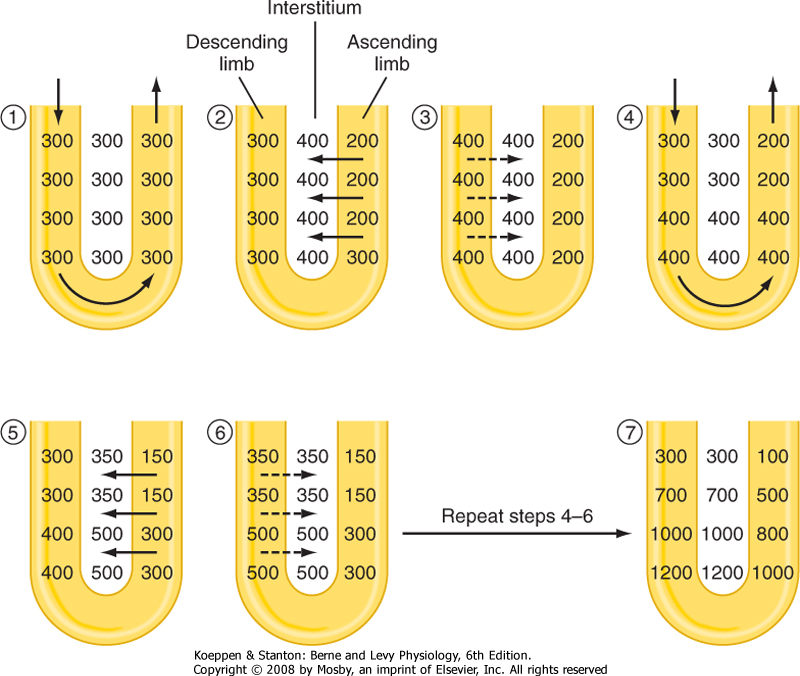
| Figure 34-5 The process of countercurrent multiplication by the loop of Henle. Initially (1), fluid in the loop of Henle and interstitium has an osmolality essentially equal to that of plasma (300 mOsm/kg H2O). Transport of solute out of the ascending limb into the interstitium represents the single effect of separating solute from water (2 and 5). The osmotic pressure gradient between the interstitium and the descending limb results in passive movement of water out of the descending limb (3 and 6). In the steady state with continuous tubular fluid flow (4), the single effect is multiplied along the length of the loop to establish an osmotic gradient, with the fluid at the bend of the loop having the highest osmolality. |
| page 603 | | | page 604 |
Next, how the kidneys excrete concentrated urine (antidiuresis) when plasma osmolality and plasma ADH levels are high is considered. The following numbers refer to those encircled in Figure 34-4, B:
- 1-4. These steps are similar to those for the production of dilute urine. An important point in understanding how concentrated urine is produced is to recognize that although reabsorption of NaCl by the ascending thin and thick limbs of the loop of Henle dilutes the tubular fluid, the reabsorbed NaCl accumulates in the medullary interstitium and raises the osmolality of this compartment. Accumulation of NaCl in the medullary interstitium is crucial for the production of urine hyperosmotic to plasma because it provides the osmotic driving force for reabsorption of water by the medullary collecting duct. The overall process by which the loop of Henle, in particular, the thick ascending limb, generates the hyperosmotic medullary interstitial gradient is termed countercurrent multiplication* (Fig. 34-5). As already noted, ADH stimulates reabsorption of NaCl by the thick
ascending limb of Henle's loop. This is thought to maintain the medullary interstitial gradient at a time when water is being added to this compartment from the medullary collecting duct, which would tend to dissipate the gradient.
- 5. Because of reabsorption of NaCl by the ascending limb of the loop of Henle, the fluid reaching the collecting duct is hypoosmotic with respect to the surrounding interstitial fluid. Thus, an osmotic gradient is established across the collecting duct. In the presence of ADH, which increases the permeability of the last half of the distal tubule and the collecting duct to water, water diffuses out of the tubule lumen, and tubule fluid osmolality increases. This diffusion of water out of the lumen of the collecting duct begins the process of urine concentration. The maximum osmolality that the fluid in the distal tubule and cortical portion of the collecting duct can attain is approximately 290 mOsm/kg H2O (i.e., the same as plasma), which is the osmolality of the interstitial fluid and plasma within the cortex of the kidney. Although the fluid at this point has the same osmolality as the fluid that entered the descending thin limb, its composition has been altered dramatically. Because of reabsorption of NaCl by the preceding nephron segments, NaCl accounts for a much smaller proportion of total tubular fluid osmolality. Instead, tubule fluid osmolality reflects the presence of urea (filtered urea plus urea added to the descending thin limb of the loop of Henle) and other solutes (e.g., K+, NH4+, and creatinine).
- 6. The osmolality of the interstitial fluid in the medulla progressively increases from the junction between the renal cortex and medulla, where it is approximately 300 mOsm/kg H2O, to the papilla, where it is approximately 1200 mOsm/kg H2O. Thus, an osmotic gradient exists between tubule fluid and interstitial fluid along the entire medullary collecting duct. In the presence of ADH, which increases the permeability of the medullary collecting duct to water, the osmolality of tubule fluid increases as water is reabsorbed. Because the initial portions of the collecting duct (cortical and outer medullary) are impermeable to urea, it remains in the tubular fluid, and its concentration increases. As already noted, in the presence of ADH, the permeability of the last portion of the medullary collecting duct (inner medullary) to urea is increased. Because the concentration of urea in the tubular fluid has been increased by reabsorption of water in the cortex and outer medulla, its concentration in tubular fluid is greater than its concentration in interstitial fluid, and some urea diffuses out of the tubule lumen into the medullary interstitium. The maximal osmolality that the fluid in the medullary collecting duct can attain is equal to that of the surrounding interstitial fluid. The major components of the tubular fluid within the medullary collecting ducts are substances that have either escaped reabsorption or have been secreted into the tubular fluid. Of these, urea is the most abundant.
- 7. The urine produced when ADH levels are elevated has an osmolality of 1200 mOsm/kg H2O and contains high concentrations of urea and other nonreabsorbed solutes. Because urea in tubular fluid equilibrates with urea in the medullary interstitial fluid, its concentration in urine is similar to that in the interstitium. Urine volume under this condition can be as low as 0.5 L/day.
|
|
Table 34-3.
Transport and Permeability Properties of Nephron Segments Involved in Urine Concentration and Dilution |
| Tubule Segment | Active Transport | Passive Permeability* | | | Effect of ADH |
| | | NaCl | Urea | H2O | |
| Loop of Henle |
| Descending thin limb | 0 | + | + | +++ | |
| Ascending thin limb | 0 | +++ | 0 | 0 | |
| Thick ascending limb | +++ | + | 0 | 0 | ↑ NaCl reabsorption |
| Distal tubule | ++ | + | 0 | 0 | ↑ H2O permeability (late portion only) |
| Collecting duct |
| Cortex | + | + | 0 | 0 | ↑ H2O permeability |
| Medulla | + | + | ++ | + | ↑ H2O and urea permeability |
*Permeability is proportional to the number of plus signs indicated: +, low permeability; + + +, high permeability; 0, impermeable.
|
| page 604 | | | page 605 |
| Movement of water across the various segments of the nephron occurs via water channels (aquaporins). The proximal tubule and the thin descending limb of Henle's loop are highly permeable to water, and these segments express high levels of AQP1 in both the apical and basolateral membrane. The vasa recta are likewise highly permeable to water and express AQP1. AQP7 and AQP8 are also expressed in the proximal tubule. As already discussed, AQP2 is responsible for ADH-regulated water movement across the apical membrane of principal cells of the late distal tubule and collecting duct, and AQP3 and AQP4 are responsible for water movement across the basolateral membrane. |
| Mice lacking the AQP1 gene have been created. These mice have a urine-concentrating defect with increased urine output. Several individuals have been found who also lack the normal AQP1 gene. Interestingly, these individuals do not have polyuria. However, when challenged by water deprivation, they are able to concentrate their urine to only approximately half of what is seen in a normal individual. |
|
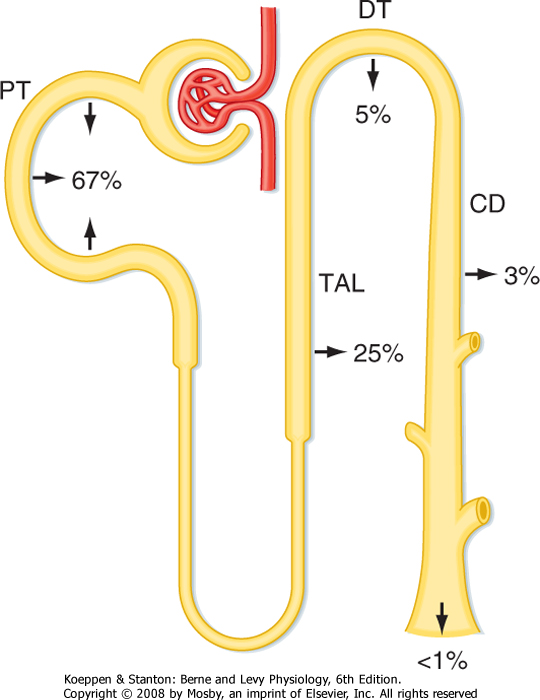
| As just described, reabsorption of water by the proximal tubule (67% of the filtered amount) and the thin descending limb of the loop of Henle (15% of the filtered amount) is essentially the same regardless of whether the urine is dilute or concentrated. As a result, a relatively constant volume of water is delivered to the distal tubule and collecting duct each day. Depending on the plasma ADH concentration, a variable portion of this water is then reabsorbed (8% to 17% of the filtered amount), with water excretion ranging from less than 1% to 10% of the filtered water.
During antidiuresis, most of the water is reabsorbed in the distal tubule and cortical and outer medullary portions of the collecting duct. Thus, a relatively small volume of fluid reaches the inner medullary collecting duct, where it is then reabsorbed. This distribution of water reabsorption along the length of the collecting duct (i.e., cortex > outer medulla > inner medulla) allows maintenance of a hyperosmotic interstitial environment in the inner medulla by minimizing the amount of water entering this compartment.
|
| As noted, the interstitial fluid of the renal medulla is critically important in concentrating urine. The osmotic pressure of interstitial fluid provides the driving force for reabsorbing water from both the descending thin limb of the loop of Henle and the collecting duct. The principal solutes of medullary interstitial fluid are NaCl and urea, but the concentration of these solutes is not uniform throughout the medulla (i.e., a gradient exists from the cortex to the papilla). Other solutes also accumulate in the medullary interstitium (e.g., NH4+ and K+), but the most abundant solutes are NaCl and urea. For simplicity, this discussion assumes that NaCl and urea are the only solutes.
|
| At the junction of the medulla with the cortex, interstitial fluid has an osmolality of approximately 300 mOsm/kg H2O, with virtually all osmoles attributable to NaCl. The concentrations of both NaCl and urea increase progressively with increasing depth into the medulla. When maximally concentrated urine is excreted, the osmolality of the medullary interstitial fluid is approximately 1200 mOsm/kg H2O at the papilla (Fig. 34-4, B). Of this value, approximately 600 mOsm/kg H2O is attributed to NaCl and 600 mOsm/kg H2O to urea. As described later, NaCl is an effective osmole in the inner medulla and thus is responsible for driving reabsorption of water from the medullary collecting duct.
|
| The medullary gradient for NaCl results from the accumulation of NaCl reabsorbed by the nephron segments in the medulla during countercurrent multiplication. The most important segment in this regard is the ascending limb (the thick limb more than the thin limb) of the loop of Henle. Accumulation of urea within the medullary interstitium is more complex and occurs most effectively when hyperosmotic urine is excreted (i.e., antidiuresis). When dilute urine is produced, especially over extended periods, the osmolality of the medullary interstitium declines (Fig. 34-4). This reduced osmolality is almost entirely caused by a decrease in the concentration of urea. This decrease reflects washout by the vasa recta (see later) and diffusion of urea from the interstitium into the tubular fluid within the medullary portion of the collecting duct. Recall that the medullary collecting duct is significantly permeable to urea even in the absence of ADH (Table 34-3).
|
| Urea is not synthesized in the kidney but is generated by the liver as a product of protein metabolism. It enters tubular fluid via glomerular filtration. As indicated in Table 34-3, the permeability of most nephron segments involved in urinary concentration and dilution to urea is relatively low. The important exception is the medullary collecting duct, which has relatively high permeability to urea that is further increased by ADH. As fluid moves along the nephron and water is reabsorbed in the collecting duct, the urea concentration in tubular fluid increases. When this urea-rich tubular fluid reaches the medullary collecting duct, where permeability to urea is not only high but also increased by ADH, urea diffuses down its concentration gradient into medullary interstitial fluid, where it accumulates. When ADH levels are elevated, the urea within the lumen of the collecting duct and the interstitium equilibrates. The resultant concentration of urea in urine is equal to that in the medullary interstitium at the papilla, or approximately 600 mOsm/kg H2O.
|
| Some of the urea within the interstitium enters the descending thin limb of the loop of Henle via the UT-A2 urea transporter. This urea is then trapped in the nephron until it again reaches the medullary collecting duct, where it can reenter the medullary interstitium. Thus, urea recycles from the interstitium to the nephron and back into the interstitium. This process of recycling facilitates the accumulation of urea in the medullary interstitium. Consequently, during antidiuresis, the concentration of urea can reach 600 mOsm/kg H2O, which is approximately half of the total medullary interstitial concentration (Fig. 34-4, B).
|
| page 605 | | | page 606 |
| Mice that lack the genes for both UT-A1 and UT-A3 have been genetically created. These mice are unable to generate a maximally hyperosmotic medullary interstitium and, as a result, can concentrate their urine to only approximately 35% of that in normal mice. This residual concentrating ability reflects reabsorption of NaCl by the ascending limb of Henle's loop and accumulation of NaCl in the medullary interstitium. |
|
| As described, the hyperosmotic medullary interstitium is essential for concentrating the tubular fluid within the collecting duct. Because reabsorption of water from the collecting duct is driven by the osmotic gradient established in the medullary interstitium, urine can never be more concentrated than that of the
interstitial fluid in the papilla. Thus, any condition that reduces medullary interstitial osmolality impairs the ability of the kidneys to maximally concentrate urine. Urea within the medullary interstitium contributes to the total osmolality of the urine. However, because the inner medullary collecting duct is highly permeable to urea, especially in the presence of ADH, urea cannot drive water reabsorption across this nephron segment (i.e., urea is an ineffective osmole). Instead, the urea in tubular fluid and the medullary interstitium equilibrates, and a small volume of urine with a high concentration of urea is excreted. This, in effect, allows the kidneys to excrete the daily urea load in a relatively small volume of urine. If urine with a high concentration of urea could not be excreted, the need to excrete the daily urea load would obligate the excretion of a much larger volume of urine. It is the medullary interstitial NaCl concentration that is responsible for reabsorbing water from the medullary collecting duct and thereby concentrating the nonurea solutes (e.g., NH4+ salts, K+ salts, creatinine) in urine.
|
| The vasa recta, the capillary networks that supply blood to the medulla, are highly permeable to solute and water (water via AQP1). As with the loop of Henle, the vasa recta form a parallel set of hairpin loops within the medulla (see Chapter 32). Not only do the vasa recta bring nutrients and oxygen to the medullary nephron segments, but more importantly, they also remove the excess water and solute that is continuously being added to the medullary interstitium by these nephron segments. The ability of the vasa recta to maintain the medullary interstitial gradient is flow dependent. A substantial increase in vasa recta blood flow dissipates the medullary gradient (i.e., washout of osmoles from the medullary interstitium). Alternatively, reduced blood flow decreases oxygen delivery to the nephron segments within the medulla. Because transport of salt and other solutes requires oxygen and ATP, reduced medullary blood flow decreases salt and solute transport by nephron segments in the medulla. As a result, the medullary interstitial osmotic gradient cannot be maintained.
|
| Assessment of Renal Diluting and Concentrating Ability
|
| The vasa recta express the UT-B urea transporter. Individuals who lack this transporter have a decreased ability to concentrate their urine. Similarly, UT-B knockout mice cannot maximally concentrate urine. Thus, in the absence of this transporter there is impaired trapping of urea in the medulla by the vasa recta. |
| Assessment of renal water handling includes measurement of urine osmolality and the volume of urine
excreted. The range of urine osmolality is 50 to 1200 mOsm/kg H2O. The corresponding range in urine volume is 18 L to as little as 0.5 L/day. These ranges are not fixed, but they vary from individual to individual and, as noted previously, depend on the amount of solute excreted.
|
| As emphasized in this chapter, the ability of the kidneys to dilute or concentrate urine requires the separation of solute and water (i.e., the single effect of the countercurrent multiplication process). This separation of solute and water in essence generates a volume of water that is "free of solute." When the urine is dilute, solute-free water is excreted from the body. When the urine is concentrated, solute-free water is returned to the body (i.e., conserved). The concept of free water clearance provides a way to calculate the amount of solute-free water generated by the kidneys, either when dilute urine is excreted or when concentrated urine is formed. As its name denotes, free water clearance is directly derived from the concept of renal clearance discussed in Chapter 32.
|
To determine free water clearance, the clearance of total solute by the kidneys must be calculated. This clearance of total solute (i.e., osmoles, whether effective or ineffective) from plasma by the kidneys is termed osmolar clearance (Cosm) and can be calculated as follows:

where Uosm is urine osmolality, V. is the urine flow rate, and Posm is the osmolality of plasma. Cosm has units of volume/unit time. Free water clearance (CH2O) is then calculated as follows:

|
| page 606 | | | page 607 |
By rearranging Equation 34-3, it should be apparent that

In other words, it is possible to partition total urine output (V.) into two hypothetical components. One component contains all the urine solutes and has an osmolality equal to that of plasma (i.e., Uosm = Posm). This volume is defined by Cosm and represents a volume from which there has been no separation of solute and
water. The second component is a volume of solute-free water (i.e., CH2O).
|
| When dilute urine is produced, the value of CH2O is positive, which indicates that solute-free water is being excreted from the body. When concentrated urine is produced, the value of CH2O is negative, which indicates that solute-free water is being retained in the body. By convention, negative CH2O values are expressed as TcH2O (tubular conservation of water).
|
| Calculation of CH2O and TcH2O can provide important information about the function of portions of the nephron involved in producing dilute and concentrated urine. Whether the kidneys excrete or reabsorb solute-free water depends on the presence of ADH. When ADH is absent or ADH levels are low, solute-free water is excreted. When ADH levels are high, solute-free water is reabsorbed.
|
The following factors are necessary for the kidneys to excrete a maximal amount of solute-free water (CH2O):
- ADH must be absent. Without ADH, the collecting duct does not reabsorb a significant amount of water.
- The tubular structures that separate solute from water (i.e., dilute the luminal fluid) must function normally. In the absence of ADH, the following nephron segments can dilute the luminal fluid:
- Ascending thin limb of Henle's loop
- Thick ascending limb of Henle's loop
- Distal tubule
- Collecting duct
|
Because of its high transport rate, the thick ascending limb is quantitatively the most important nephron segment involved in the separation of solute and water.
- 3. An adequate amount of tubular fluid must be delivered to the aforementioned nephron sites for maximal separation of solute and water. Factors that reduce delivery (e.g., decreased GFR or enhanced proximal tubule reabsorption) impair the kidneys' ability to maximally excrete solute-free water.
|
Similar requirements also apply to conservation of water by the kidneys (TcH2O). For the kidneys to conserve water maximally, the following conditions must exist:
- An adequate amount of tubular fluid must be delivered to the nephron segments that separate solute from water. The important segment in the separation of solute and water is the thick ascending limb of Henle's loop. Delivery of tubular fluid to Henle's loop depends on the GFR and proximal tubule reabsorption.
- Reabsorption of NaCl by the nephron segments must be normal; again, the most important segment is the thick ascending limb of Henle's loop.
- A hyperosmotic medullary interstitium must be present. The osmolality of interstitial fluid is maintained via reabsorption of NaCl by Henle's loop (conditions 1 and 2) and by effective accumulation of urea. Accumulation of urea in turn depends on adequate intake of dietary protein.
- Maximum levels of ADH must be present and the collecting duct must respond normally to ADH.
|
| CONTROL OF EXTRACELLULAR FLUID VOLUME AND REGULATION OF RENAL NaCL EXCRETION
|
| The major solutes in ECF are the salts of Na+. Of these, NaCl is the most abundant. Because NaCl is also the major determinant of ECF osmolality, alterations in Na+ balance are commonly assumed to disturb ECF osmolality. However, under normal circumstances such is not the case because the ADH and thirst systems maintain body fluid osmolality within a very narrow range (see earlier). For example, the addition of NaCl to ECF (without water) increases the [Na+] and osmolality of this compartment. (ICF osmolality also increases because of osmotic equilibration with ECF.) This increase in osmolality in turn stimulates thirst and release of ADH from the posterior pituitary. The increased ingestion of water in response to thirst, together with the ADH-induced decrease in excretion of water by the kidneys, quickly restores ECF osmolality to normal. However, ECF volume increases in proportion to the amount of water ingested, which in turn depends on the amount of NaCl added to ECF. Thus, in the new steady state, the addition of NaCl to ECF is equivalent to adding an isosmotic solution, and the volume of this compartment increases. Conversely, a decrease in the NaCl content of ECF lowers the volume of this compartment and is equivalent to removing an isosmotic solution.
|
| The kidneys are the major route for the excretion of NaCl from the body. Only about 10% of the Na+ lost from the body each day does so by nonrenal routes (e.g., in perspiration and feces). As such, the kidneys are critically important in regulating ECF volume. Under normal conditions, the kidneys keep ECF volume constant by adjusting the excretion of NaCl to match the amount ingested in the diet. If ingestion exceeds excretion, ECF volume increases above normal, whereas the opposite occurs if excretion exceeds ingestion.
|
| The typical diet contains approximately 140 mEq/day of Na+ (8 g of NaCl), and thus daily excretion of Na+ in urine is also about 140 mEq/day. However, the kidneys can vary the excretion of Na+ over a wide range. Excretion rates as low as 10 mEq/day can be attained when individuals are placed on a low-salt diet. Conversely, the kidneys can increase their excretion rate to more than 1000 mEq/day when challenged by the ingestion of a high-salt diet. These changes in excretion of Na+ can occur with only modest changes in ECF volume and the Na+ content of the body.
|
| page 607 | | | page 608 |
| The response of the kidneys to abrupt changes in NaCl intake typically takes several hours to several days, depending on the magnitude of the change. During this transition period, intake and excretion of Na+ are not matched as they are in the steady state.
Thus, the individual experiences either positive Na+ balance (intake > excretion) or negative Na+ balance (intake < excretion). However, by the end of the transition period, a new steady state is established, and intake once again equals excretion. Provided that the ADH and thirst systems are intact and normal, alterations in Na+ balance change the volume but not the [Na+] of the ECF. Changes in ECF volume can be monitored by measuring body weight because 1 L of ECF equals 1 kg of body weight.
|
| This section reviews the physiology of the receptors that monitor ECF volume and explains the various signals that act on the kidneys to regulate excretion of NaCl and thereby ECF volume. In addition, the responses of the various portions of the nephron to these signals are considered.
|
| Concept of Effective Circulating Volume
|
| Neuroendocrine cells in the intestine (primarily the jejunum) produce a peptide hormone called uroguanylin in response to ingestion of NaCl. A related peptide, guanylin, is also produced by the intestine (primarily the colon). These hormones have been shown to cause increased excretion of NaCl and water by the kidneys. Interestingly, both guanylin and uroguanylin are produced by the nephron (guanylin primarily in the proximal tubule and uroguanylin primarily in the collecting duct), thus suggesting a paracrine role for these peptides in the intrarenal regulation of NaCl and water transport. The actions of both uroguanylin and guanylin are mediated via activation of guanylyl cyclase (and also phospholipase A2). In the proximal tubule, guanylin and uroguanylin decrease the expression of Na+,K+-ATPase and inhibit the activity of the apical membrane Na+-H+ antiporter. In the collecting duct these peptides inhibit the K+ channel (ROMK) in the apical membrane of the principal cells, which in turn indirectly inhibits reabsorption of Na+ by changing the driving force for entry of Na+ across the apical membrane. Interestingly, mice lacking the uroguanylin gene have been found to have a blunted natriuretic response to an oral NaCl load. These mice also have increased blood pressure. Thus, uroguanylin (and guanylin) may be important hormones in regulating the renal excretion of NaCl in response to changes in NaCl intake. |
|
| As described in Chapter 2, the ECF is subdivided into two compartments: blood plasma and interstitial fluid. Plasma volume is a determinant of vascular volume and thus blood pressure and cardiac output. Maintenance of Na+ balance, and thus ECF volume, involves a complex system of sensors and effector signals that act primarily on the kidneys to regulate the excretion of NaCl. As can be appreciated from the dependency of vascular volume, blood pressure, and cardiac output on ECF volume, this complex system is designed to ensure adequate tissue perfusion. Because the primary sensors of this system are located in the large vessels
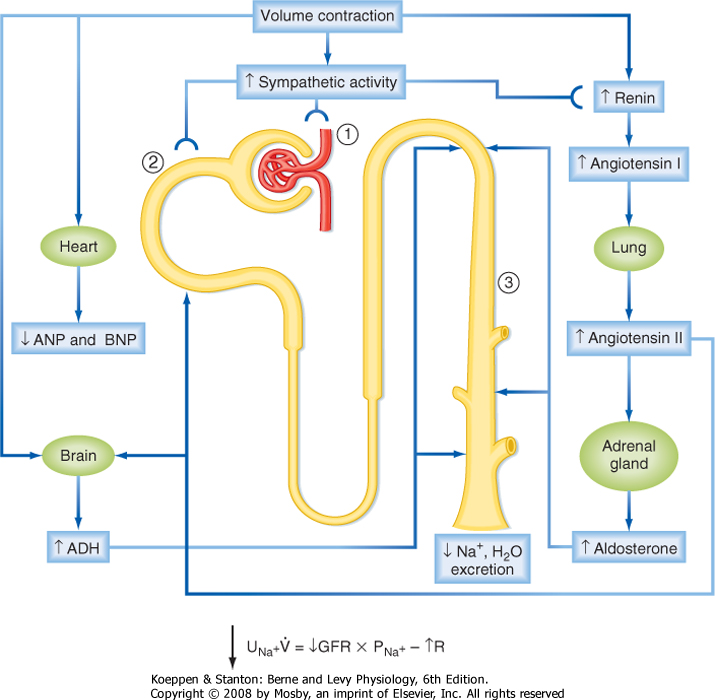
of the vascular system, changes in vascular volume, blood pressure, and cardiac output are the principal factors regulating renal NaCl excretion (see later). In a normal individual, changes in ECF volume result in parallel changes in vascular volume, blood pressure, and cardiac output. Thus, a decrease in ECF volume, a situation termed volume contraction, results in reduced vascular volume, blood pressure, and cardiac output. Conversely, an increase in ECF volume, a situation termed volume expansion, results in increased vascular volume, blood pressure, and cardiac output. The extent to which these cardiovascular parameters change is dependent on the degree of volume contraction or expansion and the effectiveness of cardiovascular reflex mechanisms (see Chapters 18 and 19). When a person is in negative Na+ balance, ECF volume is decreased and renal NaCl excretion is reduced. Conversely, with positive Na+ balance there is an increase in ECF volume, which results in enhanced renal NaCl excretion (i.e., natriuresis).
|
| However, in some pathological conditions (e.g., congestive heart failure, hepatic cirrhosis), the renal excretion of NaCl does not reflect the ECF volume. In both these situations ECF volume is increased. However, instead of increased renal NaCl excretion, as would be expected, there is a reduction in the renal excretion of NaCl. To explain renal Na+ handling in these situations, it is necessary to understand the concept of effective circulating volume (ECV). Unlike ECF, ECV is not a measurable and distinct body fluid compartment. ECV refers to the portion of the ECF that is contained within the vascular system and is "effectively" perfusing the tissues (effective blood volume and effective arterial blood volume are other commonly used terms). More specifically, ECV reflects the activity of volume sensors located in the vascular system (see later).
|
| In normal individuals, ECV varies directly with the volume of the ECF and in particular the volume of the vascular system (arterial and venous), arterial blood pressure, and cardiac output. However, as noted, this is not the case in certain pathological conditions. The remaining sections of this chapter examine the relationship between ECF volume and renal NaCl excretion in normal adults, where changes in ECV and ECF volume occur in parallel.
|
|
Table 34-4.
Volume and Na+ Sensors |
| I. Vascular |
| A. Low pressure |
| 1. Cardiac atria |
| 2. Pulmonary vasculature |
| B. High pressure |
| 1. Carotid sinus |
| 2. Aortic arch |
| 3. Juxtaglomerular apparatus of the kidney |
| II. Central nervous system |
| III. Hepatic |
| page 608 | | | page 609 |
| Patients with congestive heart failure frequently have an increase in ECF volume that is manifested as increased plasma volume and accumulation of interstitial fluid in the lungs (pulmonary edema) and peripheral tissues (generalized edema). This excess fluid is the result of NaCl and water retention by the kidneys. The kidneys' response (i.e., retention of NaCl and water) is paradoxical because ECF volume is increased. However, this fluid is not in the vascular system but in the interstitial fluid compartment. In addition, blood pressure and cardiac output may be reduced because of poor cardiac performance. Therefore, the sensors located in the vascular system respond as they do in ECF volume contraction and cause retention of NaCl and water by the kidneys. In this situation, ECV, as monitored by volume sensors, is decreased. |
| Large volumes of fluid accumulate in the peritoneal cavity of patients with advanced hepatic cirrhosis. This fluid, called ascites, is a component of ECF and results from retention of NaCl and water by the kidneys. Again, the response of the kidneys in this situation seems paradoxical if only ECF volume is considered. With advanced hepatic cirrhosis, blood pools in the splanchnic circulation (i.e., the damaged liver impedes the drainage of blood from the splanchnic circulation via the portal vein). Thus, volume and pressure are reduced in the portions of the vascular system in which the sensors are found, but venous pressure in the portal system increases, which enhances fluid transudation into the peritoneal cavity. Hence, the kidneys respond as they would during ECF volume contraction: retention of NaCl and water and accumulation of ascites fluid. As with congestive heart failure, ECV in cirrhosis with ascites is decreased. |
|
| ECF volume (or ECV) is monitored by multiple sensors (Table 34-4). A number of the sensors are located in
the vascular system, and they monitor its fullness and pressure. These receptors are typically called volume receptors, or because they respond to pressure-induced stretch of the walls of the receptor (e.g., blood vessels or cardiac atria), they are also referred to as baroreceptors (see earlier). The sensors within the liver and central nervous system (CNS) are less well understood and do not seem to be as important as the vascular sensors in monitoring ECF volume.
|
| Vascular Low-Pressure Volume Sensors
|
| Volume sensors (i.e., baroreceptors) are located within the walls of the cardiac atria, right ventricle, and large pulmonary vessels, and they respond to distention of these structures (see also Chapters 18 and 19). Because the low-pressure side of the circulatory system has high compliance, these sensors respond mainly to "fullness" of the vascular system. These baroreceptors send signals to the brainstem via afferent fibers in the glossopharyngeal and vagus nerves. The activity of these sensors modulates both sympathetic nerve outflow and ADH secretion. For example, a decrease in filling of the pulmonary vessels and cardiac atria increases sympathetic nerve activity and stimulates secretion of ADH. Conversely, distention of these structures decreases sympathetic nerve activity. In general, 5% to 10% changes in blood volume and pressure are necessary to evoke a response.
|
| The cardiac atria possess an additional mechanism related to control of renal NaCl excretion. The myocytes of the atria synthesize and store a peptide hormone. This hormone, termed atrial natriuretic peptide (ANP), is released when the atria are distended, which via mechanisms outlined later in this chapter, reduces blood pressure and increases the excretion of NaCl and water by the kidneys. The ventricles of the heart also produce a natriuretic peptide termed brain natriuretic peptide (BNP), so named because it was first isolated from the brain. Like ANP, BNP is released from ventricular myocytes by distention of the ventricles. Its actions are similar to those of ANP.
|
| Vascular High-Pressure Volume Sensors
|
| Baroreceptors are also present in the arterial side of the circulatory system, located in the walls of the aortic arch, carotid sinus, and afferent arterioles of the kidneys. The aortic arch and carotid baroreceptors send input to the brainstem via afferent fibers in the glossopharyngeal and vagus nerves. The response to this input alters sympathetic outflow and ADH secretion. Thus, a decrease in blood pressure increases sympathetic nerve activity and ADH secretion. An increase in pressure tends to reduce sympathetic nerve activity (and activate parasympathetic nerve activity). The sensitivity of the high-pressure baroreceptors is similar to that in the low-pressure side of the vascular system; 5% to 10% changes in pressure are needed to evoke a response.
|
| The juxtaglomerular apparatus of the kidneys (see Chapter 32), particularly the afferent arteriole, responds directly to changes in pressure. If perfusion pressure in the afferent arteriole is reduced, renin is released from myocytes. Secretion of renin is suppressed when perfusion pressure is increased. As described later in this chapter, renin determines blood levels of angiotensin II and aldosterone, both of which play an important role in regulating renal NaCl excretion.
|
| Of the two classes of baroreceptors, those on the high-pressure side of the vascular system appear to be more important in influencing sympathetic tone and ADH secretion. For example, patients with congestive heart failure often have increased vascular volume with dilation of the atria and ventricles. This would be expected to decrease sympathetic tone and inhibit ADH secretion via the low-pressure baroreceptors. However, sympathetic tone is often increased and ADH secretion stimulated in these patients (the renin-angiotensin-aldosterone system is also activated). This reflects the activity of the high-pressure baroreceptors in response to reduced blood pressure and cardiac output secondary to the failing heart (i.e., the high-pressure baroreceptors detect a reduced ECV).
|
| page 609 | | | page 610 |
| Constriction of a renal artery by an atherosclerotic plaque, for example, reduces perfusion pressure to that kidney. This reduced perfusion pressure is sensed by the afferent arteriole of the juxtaglomerular apparatus and results in the secretion of renin. The elevated renin levels increase the production of angiotensin II, which in turn increases systemic blood pressure via its vasoconstrictor effect on arterioles throughout the vascular system. The increased systemic blood pressure is sensed by the juxtaglomerular apparatus of the contralateral kidney (i.e., the kidney without stenosis of its renal artery), and renin secretion from that kidney is suppressed. In addition, the high levels of angiotensin II act to inhibit renin secretion by the contralateral kidney (negative feedback). Treatment of patients with constricted renal arteries includes surgical repair of the stenotic artery, administration of angiotensin II receptor blockers, or administration of an inhibitor of angiotensin-converting enzyme (ACE). The ACE inhibitor blocks the conversion of angiotensin I to angiotensin II. |
|
| Though not as important as the vascular sensors, the liver also contains volume sensors that can modulate renal NaCl excretion. One type of hepatic sensor responds to pressure within the hepatic vasculature and therefore functions in a manner similar to the low- and high-pressure baroreceptors. A second type of sensor also appears to exist in the liver. This sensor responds to the [Na+] of the portal blood entering the liver. Afferent signals from both types of sensors are sent to the same area of the brainstem where afferent fibers from both the low- and high-pressure baroreceptors converge. Increased pressure within the hepatic vasculature or an increase in portal blood [Na+] results in a decrease in efferent sympathetic nerve activity.* As described later, this decreased sympathetic nerve activity leads to an increase in renal NaCl excretion.
|
| Central Nervous System Na+ Sensors
|
|
Table 34-5.
Signals Involved in Control of Renal NaCl and Water Excretion |
| Renal Sympathetic Nerves (↑ Activity: ↓ NaCl Excretion) |
| ↓ GFR |
| ↑ Renin secretion |
| ↑ Na+ reabsorption along the nephron |
| Renin-Angiotensin-Aldosterone (↑ Secretion: ↓ NaCl Excretion) |
| ↑ Angiotensin II stimulates reabsorption of Na+ along the nephron |
| ↑ Aldosterone stimulates Na+ reabsorption in the thick ascending limb of Henle's loop, distal tubule, and collecting duct |
| ↑ Angiotensin II stimulates secretion of ADH |
| Natriuretic Peptides: ANP, BNP, and Urodilatin (↑ Secretion: ↑ NaCl Excretion) |
| ↑ GFR |
| ↓ Renin secretion |
| ↓ Aldosterone secretion (indirect via ↓ in angiotensin II and direct on the adrenal gland) |
| ↓ NaCl and water reabsorption by the collecting duct |
| ↓ ADH secretion and inhibition of ADH action on the distal tubule and collecting duct |
| ADH (↑ Secretion: ↓ H2O Excretion) |
| ↑ H2O reabsorption by the distal tubule and collecting duct |
| Like the hepatic sensors, the CNS sensors do not appear to be as important as the vascular sensors in monitoring ECF volume and controlling renal NaCl excretion. Nevertheless, alterations in the [Na+] of blood carried to the brain in the carotid arteries or the [Na+] of cerebrospinal fluid (CSF) modulate renal NaCl excretion. For example, if the [Na+] in either carotid artery blood or CSF is increased, there is a decrease in renal sympathetic nerve activity, which in turn leads to an increase in renal NaCl excretion. The hypothalamus appears to be the site where these sensors are located. Angiotensin II and natriuretic peptides are generated in the hypothalamus. These locally generated signals, together with systemically generated
angiotensin II and natriuretic peptides, appear to play a role in modulating the CNS Na+-sensing system.
|
| Of the volume and Na+ sensors just described, those located in the vascular system are better understood. Moreover, their function in health and disease explains quite effectively the regulation of renal NaCl excretion. Therefore, the remainder of this chapter will focus on the vascular volume sensors (i.e., baroreceptors) and their role in regulating renal NaCl excretion.
|
| When the vascular volume sensors have detected a change in ECF volume, they send signals to the kidneys, which results in an appropriate adjustment in excretion of NaCl and water. Accordingly, when ECF volume is expanded, renal NaCl and water excretion is increased. Conversely, when ECF volume is contracted, renal NaCl and water excretion is reduced. The signals involved in coupling the volume sensors to the kidneys are both neural and hormonal. These signals are summarized in Table 34-5, as are their effects on renal NaCl and water excretion.
|
| page 610 | | | page 611 |
As described in Chapter 33, sympathetic nerve fibers innervate the afferent and efferent arterioles of the glomerulus, as well as the cells of the nephron. With ECF volume contraction, activation of the low- and high-pressure vascular baroreceptors results in stimulation of sympathetic nerve activity, including fibers innervating the kidneys. This has the following effects:
- The afferent and efferent arterioles are constricted (mediated by α-adrenergic receptors). This vasoconstriction (the effect is greater on the afferent arteriole) decreases hydrostatic pressure within the glomerular capillary lumen, which results in a decrease in GFR. With this decrease in GFR, the filtered load of Na+ to the nephrons is reduced.
- Renin secretion is stimulated by cells of the afferent arterioles (mediated by β-adrenergic receptors). As described later, renin ultimately increases the circulating levels of angiotensin II and aldosterone, both of which stimulate Na+ reabsorption by the nephron.
- NaCl reabsorption along the nephron is directly stimulated (mediated by α-adrenergic receptors on cells of the nephron). Because of the large amount of Na+ reabsorbed by the proximal tubule, the effect of increased sympathetic nerve activity is quantitatively most important for this segment.
|
| As a result of these actions, increased renal sympathetic nerve activity decreases excretion of NaCl, an adaptive response that works to restore ECF volume to normal, a state termed euvolemia. With ECF volume expansion, renal sympathetic nerve activity is reduced. This generally reverses the effects just described.
|
| Renin-Angiotensin-Aldosterone System
|
| Recently, a new "renal hormone" was discovered: a flavin adenine dinucleotide-dependent amine oxidase named renalase. Renalase is similar in structure to monoamine oxidase and metabolizes catecholamines (e.g., dopamine, epinephrine, and norepinephrine). Other tissues also express renalase (e.g., skeletal muscle, heart, small intestine), but the kidneys secrete the enzyme into the circulation. Because individuals with chronic renal failure have very low levels of renalase in their plasma, the kidney is probably the primary source of the circulating enzyme. In experimental animals, infusion of renalase decreases blood pressure and heart contractility. Although the precise role of renalase in regulation of cardiovascular function and blood pressure is not known, it may be important in modulating the effect of the sympathetic nervous system, especially the effects of sympathetic nerves on the kidney. |
|
| Although many tissues express renin (e.g., brain, heart, adrenal gland), the primary source of circulating renin is the kidneys. Renin is secreted by juxtaglomerular cells located in the afferent arteriole. At the cellular level, secretion of renin is mediated by the fusion of renin-containing granules with the luminal membrane of the cell. This process is stimulated by a decrease in intracellular [Ca++], a response opposite that of most secretory cells, where secretion is stimulated by an increase in intracellular [Ca++]. It is also stimulated by an increase in intracellular [cAMP]. Thus, anything that increases intracellular [Ca++] will inhibit renin secretion. This would include stretch of the afferent arteriole (myogenic control of renin secretion), angiotensin II (i.e., feedback inhibition), and endothelin. Conversely, anything that increases intracellular [cAMP] will stimulate renin secretion. This would include norepinephrine acting via β-adrenergic receptors and prostaglandin E2. Increases in intracellular [cGMP] have been shown to stimulate renin secretion in some situations and to inhibit secretion in others. Importantly, two substances that increase intracellular [cGMP] are ANP and nitric oxide. Both inhibit renin secretion. |
| Control of renin secretion by the macula densa is complex and appears to involve several paracrine factors. For example, when delivery of NaCl to the macula densa is increased, ATP (and perhaps also adenosine) is released across the basolateral membrane. Binding of ATP to receptors on extraglomerular mesangial cells results in an increase in intracellular [Ca++]. Because mesangial cells are coupled to juxtaglomerular cells by gap junctions, the intracellular [Ca++] in juxtaglomerular cells also increases, and renin secretion is suppressed. This increase in the intracellular [Ca++] of mesangial cells also increases the intracellular [Ca++] of vascular smooth muscle cells of the afferent arteriole (again via gap junctions), thereby resulting in constriction and thus a decrease in GFR (see also Chapter 32). When delivery of NaCl to the macula densa is decreased, release of ATP and adenosine is suppressed, and the intracellular [Ca++] of mesangial, juxtaglomerular, and vascular smooth muscle cells decreases. This stimulates secretion of renin by the juxtaglomerular cells, and the afferent arteriole dilates. In addition, with decreased delivery of NaCl, macula densa cells release prostaglandin E2, which also stimulates renin secretion and causes afferent arteriole dilation. |
|
| page 611 | | | page 612 |
Cells in the afferent arterioles (juxtaglomerular cells) are the site of synthesis, storage, and release of the proteolytic enzyme renin. Three factors are important in stimulating renin secretion:
- Perfusion pressure. The afferent arteriole behaves as a high-pressure baroreceptor. When perfusion pressure to the kidneys is reduced, renin secretion is stimulated. Conversely, an increase in perfusion pressure inhibits release of renin.
- Sympathetic nerve activity. Activation of the sympathetic nerve fibers that innervate the afferent arterioles increases renin secretion (mediated by β-adrenergic receptors). Renin secretion is decreased as renal sympathetic nerve activity is decreased.
- Delivery of NaCl to the macula densa. Delivery of NaCl to the macula densa regulates the GFR by a process termed tubuloglomerular feedback (see Chapter 32). In addition, the macula densa plays a role in renin secretion. When delivery of NaCl to the macula densa is decreased, renin secretion is enhanced. Conversely, an increase in NaCl delivery inhibits renin secretion. It is likely that macula densa-mediated renin secretion helps maintain systemic arterial pressure under conditions of reduced vascular volume. For example, when vascular volume is reduced, perfusion of body tissues (including the kidneys) decreases. This in turn decreases the GFR and the filtered load of NaCl. The reduced delivery
of NaCl to the macula densa then stimulates secretion of renin, which acts through angiotensin II (a potent vasoconstrictor) to increase blood pressure and thereby maintain tissue perfusion.
|

|
| Figure 34-6 Schematic representation of the essential components of the renin-angiotensin-aldosterone system. Activation of this system results in a decrease in the excretion of Na+ and water by the kidneys. Note: Angiotensin I is converted to angiotensin II by ACE, which is present on all vascular endothelial cells. As shown, the endothelial cells within the lungs play a significant role in this conversion process. |
Figure 34-6 summarizes the essential components of the renin-angiotensin-aldosterone system. Renin alone does not have a physiological function; it functions solely as a proteolytic enzyme. Its substrate is a circulating protein, angiotensinogen, which is produced by the liver. Angiotensinogen is cleaved by renin to yield a 10-amino acid peptide, angiotensin I. Angiotensin I also has no known physiological function, and it is further cleaved to an eight-amino acid peptide, angiotensin II, by a converting enzyme (ACE) found on the surface of vascular endothelial cells. (Pulmonary and renal endothelial cells are important sites for the conversion of angiotensin I to angiotensin II.) ACE also degrades bradykinin, a potent vasodilator.* Angiotensin II has several important physiological functions, including
- Stimulation of aldosterone secretion by the adrenal cortex.
- Arteriolar vasoconstriction, which increases blood pressure.
- Stimulation of ADH secretion and thirst.
- Enhancement of NaCl reabsorption by the proximal tubule, thick ascending limb of Henle's loop, the distal tubule, and the collecting duct. Of these segments the effect on the proximal tubule is quantitatively the largest.
|
| Angiotensin II is an important secretagogue for aldosterone. An increase in plasma [K+] is the other important stimulus for aldosterone secretion (see Chapter 35). Aldosterone is a steroid hormone produced by the glomerulosa cells of the adrenal cortex. It acts in a number of ways on the kidneys (see also Chapters 35 and 36). With regard to the regulation of ECF volume, aldosterone reduces excretion of NaCl by stimulating its reabsorption by the thick ascending limb of the loop of Henle, distal tubule, and collecting duct. The effect of aldosterone on renal NaCl excretion depends mainly on its ability to stimulate reabsorption of Na+ in the distal tubule, as well as the collecting duct. (Note: These segments are often referred to collectively as the aldosterone-sensitive distal nephron).
|
| Aldosterone has many cellular actions in responsive cells (see also Chapter 33). Importantly, it increases the abundance of the apical membrane Na+-Cl- symporter in cells of the early portion of the distal tubule and the abundance of the Na+ channel (ENaC) in the apical membrane of principal cells in the late portion of the distal tubule and collecting duct (the activity of Na+ channels is also increased). These actions of aldosterone increase entry of Na+ into cells across the apical membrane. Extrusion of Na+ from cells across the basolateral membrane occurs by Na+,K+-ATPase, the abundance of which is also increased by aldosterone. Thus, aldosterone increases reabsorption of Na+ from tubular fluid by distal nephron segments, whereas reduced levels of aldosterone decrease the amount of Na+ reabsorbed by these segments.
|
| page 612 | | | page 613 |
| The response to aldosterone has two phases. In the initial phase, which occurs within minutes, reabsorption of Na+ in the aldosterone-sensitive distal nephron increases without changes in transporter abundance. This phase reflects the activation of existing transporters, as well as inhibition of the cell's normal process of removal and recycling of membrane transport proteins (see Chapter 1). By slowing the retrieval process, more transporters are retained in the membrane, thereby increasing entry of Na+ into the cell across the apical membrane (see Chapter 33 for details). In the second phase, which occurs with a lag period of several hours, there is increased synthesis of key Na+ transport proteins, including the Na+-Cl- symporter (NCC/TSC) in the early distal tubule, the α subunit of the Na+ channel (ENaC)* in the late distal tubule and collecting duct, and the α subunit of Na+,K+-ATPase in these same segments. |
|
| Diseases of the adrenal cortex can alter aldosterone levels and thereby impair the ability of the kidneys to maintain Na+ balance and euvolemia. With decreased secretion of aldosterone (hypoaldosteronism), reabsorption of Na+, mainly by the aldosterone-sensitive distal nephron, is reduced, and NaCl is lost in urine. Because urinary NaCl loss can exceed the amount of NaCl ingested in the diet, negative Na+ balance ensues, and ECF volume decreases. In response to the ECF volume contraction, sympathetic tone is increased, and levels of renin, angiotensin II, and ADH are elevated. With increased aldosterone secretion (hyperaldosteronism), the effects are the opposite: Na+ reabsorption by the aldosterone-sensitive distal nephron is enhanced, and excretion of NaCl is reduced. Consequently, ECF volume is increased, sympathetic tone is decreased, and levels of renin, angiotensin II, and ADH are decreased. As described later, ANP and BNP levels are also elevated in this setting. |
|
| As noted, aldosterone also enhances reabsorption of Na+ by cells of the thick ascending limb of the loop of Henle, though to a lesser degree than in the aldosterone-sensitive distal nephron. This action probably reflects increased entry of Na+ into the cell across the apical membrane (most likely by the apical membrane 1Na+-1K+-2Cl- symporter) and increased extrusion from the cell by the basolateral membrane Na+,K+-ATPase.
|
| As summarized in Table 34-5, activation of the renin-angiotensin-aldosterone system, as occurs with ECF volume depletion, decreases the excretion of NaCl by the kidneys. This system is suppressed with ECF volume expansion, and renal NaCl excretion is therefore enhanced.
|
| The body produces a number of substances that act on the kidneys to increase Na+ excretion.* Of these, natriuretic peptides produced by the heart and kidneys are best understood and will be the focus of the following discussion.
|
The heart produces two natriuretic peptides. Atrial myocytes primarily produce and store the peptide hormone ANP, and ventricular myocytes primarily produce and store BNP. Both peptides are secreted when the heart dilates (i.e., during volume expansion and with heart failure), and they act to relax vascular smooth muscle and promote excretion of NaCl and water by the kidneys. The kidneys also produce a related natriuretic peptide termed urodilatin. Its actions are limited to promoting NaCl excretion by the kidneys. In general, the actions of these natriuretic peptides, as they relate to renal NaCl and water excretion, antagonize those of the renin-angiotensin-aldosterone system. These actions include
- Vasodilation of the afferent and vasoconstriction of the efferent arterioles of the glomerulus. This increases GFR and the filtered load of Na+.
- Inhibition of renin secretion by the afferent arterioles.
- Inhibition of aldosterone secretion by the glomerulosa cells of the adrenal cortex. This occurs via two mechanisms: (a) inhibition of renin secretion by the juxtaglomerular cells and consequently a reduction in angiotensin II-induced aldosterone secretion and (b) direct inhibition of aldosterone secretion by the glomerulosa cells of the adrenal cortex.
- Inhibition of NaCl reabsorption by the collecting duct, which is also caused in part by reduced levels of aldosterone. However, the natriuretic peptides also act directly on the collecting duct cells. Through the second messenger cGMP, natriuretic peptides inhibit cation channels in the apical membrane and thereby decrease reabsorption of Na+. This effect occurs predominantly in the medullary portion of the collecting duct.
- Inhibition of ADH secretion by the posterior pituitary and ADH action on the collecting duct. These effects decrease water reabsorption by the collecting duct and thus increase excretion of water in urine.
|
| page 613 | | | page 614 |
| The foregoing effects of natriuretic peptides increase the excretion of NaCl and water by the kidneys. Hypothetically, a reduction in circulating levels of these peptides would be expected to decrease NaCl and
water excretion, but convincing evidence for this effect has not been reported.
|
| As discussed previously, a decreased ECF volume stimulates secretion of ADH by the posterior pituitary. The elevated levels of ADH decrease water excretion by the kidneys, which serves to reestablish euvolemia.
|
| Control of NaCl Excretion during Euvolemia
|
| Maintenance of Na+ balance and therefore euvolemia requires precise matching of the amount of NaCl ingested with the amount excreted from the body. As already noted, the kidneys are the major route for excretion of NaCl. Accordingly, in a euvolemic individual we can equate daily urine NaCl excretion with daily NaCl intake.
|
| The amount of NaCl excreted by the kidneys can vary widely. Under conditions of salt restriction (i.e., low-NaCl diet), virtually no Na+ appears in the urine. Conversely, in individuals who ingest large quantities of NaCl, renal Na+ excretion can exceed 1000 mEq/day. The kidneys require several days to respond maximally to variations in dietary NaCl intake. During the transition period, excretion does not match intake, and the individual is in either positive (intake > excretion) or negative (intake < excretion) Na+ balance. When Na+ balance is altered during these transition periods, ECF volume changes in parallel. Water excretion, regulated via the ADH system, is also adjusted to keep plasma osmolality constant, and an isosmotic change in ECF volume results. Thus, with positive Na+ balance, ECF volume expands (detected as an acute increase in body weight), whereas with negative Na+ balance, ECF volume contracts (detected as an acute decrease in body weight). Ultimately, renal excretion reaches a new steady state and NaCl excretion once again is matched to intake. The time course for adjustment of renal NaCl excretion varies (hours to days) and depends on the magnitude of the change in NaCl intake. Adaptation to large changes in NaCl intake requires a longer time than adaptation to small changes in intake.
|
| The general features of Na+ handling along the nephron must be understood to comprehend how renal Na+ excretion is regulated. (See Chapter 33 for the cellular mechanisms of Na+ transport along the nephron.) Most (67%) of the filtered load of Na+ is reabsorbed by the proximal tubule. An additional 25% is reabsorbed by the thick ascending limb of the loop of Henle and the remainder by the distal tubule and collecting duct (Fig. 34-7).
|
In a normal adult, the filtered load of Na+ is approximately 25,000 mEq/day.
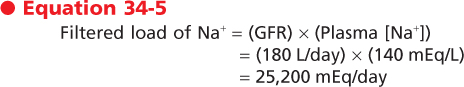
|
| With a typical diet, less than 1% of this filtered load is excreted in urine (approximately 140 mEq/day).* Because of the large filtered load of Na+, small changes in Na+ reabsorption by the nephron can profoundly affect Na+ balance and, thus, ECF volume. For example, an increase in Na+ excretion from 1% to 3% of the filtered load represents an additional loss of approximately 500 mEq/day of Na+. Because [Na+] in ECF is 140 mEq/L, such an Na+ loss would decrease ECF volume by more than 3 L (i.e., water excretion would parallel the loss of Na+ to maintain body fluid osmolality constant: 500 mEq/day/140 mEq/L = 3.6 L/day of fluid loss). Such fluid loss in a 70-kg individual would represent a 26% decrease in ECF volume.
|
In euvolemic subjects, the nephron segments distal to the loop of Henle (distal tubule and collecting duct) are the main nephron segment where Na+ reabsorption is adjusted to maintain excretion at a level appropriate for dietary intake. However, this does not mean that the other portions of the nephron are not involved in this process. Because the reabsorptive capacity of the distal tubule and collecting duct is limited, these other portions of the nephron (i.e., proximal tubule and loop of Henle) must reabsorb the bulk of the filtered load of Na+. Thus, during euvolemia, Na+ handling by the nephron can be explained by two general processes:
- Na+ reabsorption by the proximal tubule and loop of Henle is regulated so that a relatively constant portion of the filtered load of Na+ is delivered to the distal tubule. The combined action of the proximal tubule and loop of Henle reabsorbs approximately 92% of the filtered load of Na+, and thus 8% of the filtered load is delivered to the distal tubule.
- Reabsorption of this remaining portion of the filtered load of Na+ by the distal tubule and collecting duct is regulated so that the amount of Na+ excreted in urine matches the amount ingested in the diet. Thus, these later nephron segments make final adjustments in Na+ excretion to maintain the euvolemic state.
|
| Mechanisms for Maintaining Delivery of NaCl to the Distal Tubule Constant
|
| A number of mechanisms maintain a constant delivery of Na+ to the beginning of the distal tubule. These processes are autoregulation of the GFR (and thus the filtered load of Na+), glomerulotubular balance, and dependency of Na+ reabsorption by the loop of Henle on load.
|
| Autoregulation of the GFR (see Chapter 32) allows maintenance of a relatively constant filtration rate over a wide range of perfusion pressures. Because the filtration rate is constant, the filtered load of Na+ is also constant.
|
| page 614 | | | page 615 |
| Despite the autoregulatory control of GFR, small variations occur. If these changes are not compensated for by an appropriate adjustment in Na+ reabsorption by the nephron, excretion of Na+ would
change markedly. Fortunately, Na+ reabsorption in the euvolemic state, especially by the proximal tubule, changes in parallel with changes in GFR. This phenomenon is termed glomerulotubular balance. Thus, if the GFR increases, the amount of Na+ reabsorbed by the proximal tubule also increases. The opposite occurs if the GFR decreases (see Chapter 33 for a more detailed description of glomerulotubular balance).
|
| The final mechanism that helps maintain constant delivery of Na+ to the beginning of the collecting duct involves the ability of the loop of Henle to increase its reabsorptive rate in response to increased delivery of Na+.
|
| Regulation of NaCl Reabsorption by the Distal Tubule and Collecting Duct
|
| When delivery of Na+ is constant, small adjustments in Na+ reabsorption by the distal tubule and, to a lesser degree, by the collecting duct are sufficient to balance excretion with intake. As already noted, as little as a 2% change in fractional Na+ excretion produces more than a 3-L change in ECF volume. Aldosterone is the primary regulator of Na+ reabsorption by the distal tubule and collecting duct and, thus, the primary regulator of Na+ excretion under this condition. When aldosterone levels are elevated, reabsorption of Na+ by these segments is increased (excretion decreased). When aldosterone levels are decreased, reabsorption of Na+ is decreased (excretion increased).
|
| In addition to aldosterone, a number of other factors, including natriuretic peptides, prostaglandins, uroguanylin, adrenomedullin, and sympathetic nerves, alter reabsorption of Na+ by the distal tubule and collecting duct. However, the relative effects of these other factors on regulation of Na+ reabsorption by these segments during euvolemia are unclear.
|
| As long as variations in the dietary intake of NaCl are minor, the mechanisms previously described can regulate renal Na+ excretion appropriately and thereby maintain euvolemia. However, these mechanisms cannot effectively handle significant changes in NaCl intake. When NaCl intake changes significantly, ECF volume expansion or ECF volume contraction occurs. In such cases, additional factors act on the kidneys to adjust Na+ excretion and thereby reestablish the euvolemic state.
|
| Control of NaCl Excretion with Volume Expansion
|
During ECF volume expansion, the high-pressure and low-pressure vascular volume sensors send signals to the kidneys that result in increased excretion of NaCl and water. The signals acting on the kidneys include
- Decreased activity of the renal sympathetic nerves
- Release of ANP and BNP from the heart and urodilatin from the kidneys
- Inhibition of ADH secretion from the posterior pituitary and decreased ADH action on the collecting duct
- Decreased renin secretion and thus decreased production of angiotensin II
- Decreased aldosterone secretion, which is caused by reduced angiotensin II levels, and elevated natriuretic peptide levels
|
| Figure 34-7 Segmental Na+ reabsorption. The percentage of the filtered load of Na+ reabsorbed by each nephron segment is indicated. CD, cortical collecting duct; DT, distal tubule; PT, proximal tubule; TAL, thick ascending limb. |
| page 615 | | | page 616 |
The integrated response of the nephron to these signals is illustrated in Figure 34-8. Three general responses to ECF volume expansion occur (the numbers correlate with those encircled in Fig. 34-8):
- The GFR increases. The GFR increases mainly as a result of the decrease in sympathetic nerve activity. Sympathetic fibers innervate the afferent and efferent arterioles of the glomerulus and control their diameter. Decreased sympathetic nerve activity leads to arteriolar dilation. Because the effect appears to be greater on the afferent arterioles, hydrostatic pressure within the glomerular capillary is increased, thereby increasing the GFR. Because renal plasma flow increases to a greater degree than the GFR does, the filtration fraction decreases. Natriuretic peptides also increase GFR by dilating the afferent and constricting the efferent arterioles. Thus, the increased natriuretic peptide levels that occur during ECF volume expansion contribute to this response. With the increase in GFR, the filtered load of Na+ increases.
- Reabsorption of Na+ decreases in the proximal tubule and loop of Henle. Several mechanisms may act to reduce Na+ reabsorption by the proximal tubule, but the precise role of each of these mechanisms remains controversial. Because activation of the sympathetic nerve fibers that innervate this nephron segment stimulates reabsorption of Na+, the decreased sympathetic nerve activity that results from ECF volume expansion decreases Na+ reabsorption. In addition, angiotensin II directly stimulates reabsorption of Na+ by the proximal tubule. Because angiotensin II levels are also reduced under this condition, proximal tubule Na+
reabsorption decreases as a result. The increased hydrostatic pressure within the glomerular capillaries also tends to increase the hydrostatic pressure within the peritubular capillaries. In addition, the decrease in filtration fraction reduces peritubular oncotic pressure. These alterations in capillary Starling forces reduce the absorption of solute (e.g., NaCl) and water from the lateral intercellular space and thus reduce tubular reabsorption (see Chapter 33 for a complete description of this mechanism). Both the increase in the filtered load and the decrease in NaCl reabsorption by the proximal tubule result in the delivery of more NaCl to the loop of Henle. Because activation of sympathetic nerves and aldosterone stimulates reabsorption of NaCl by the loop of Henle, the reduced nerve activity and low aldosterone levels that occur with ECF volume expansion serve to reduce NaCl reabsorption by this nephron segment. Thus, the fraction of the filtered load delivered to the distal tubule is increased.
- Na+ reabsorption decreases in the distal tubule and collecting duct. As noted, the amount of Na+ delivered to the distal tubule exceeds that observed in the euvolemic state (i.e., the amount of Na+ delivered to the distal tubule varies in proportion to the degree of ECF volume expansion). This increased load of Na+ overwhelms the reabsorptive capacity of the distal tubule and the collecting duct, and this capacity is even further impaired by the actions of natriuretic peptides and by the decrease in circulating levels of aldosterone.
|
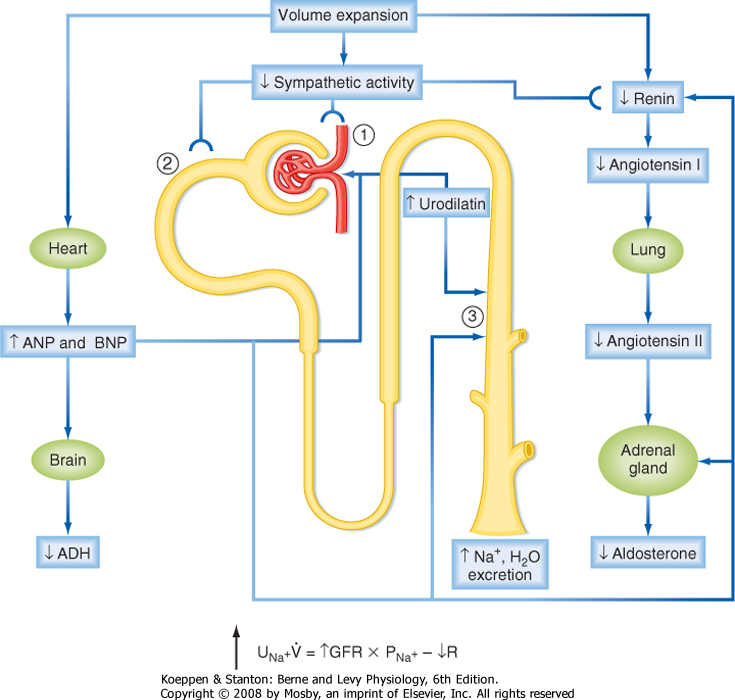
|
| Figure 34-8 Integrated response to ECF volume expansion. Numbers refer to the description of the response in the text. PNa+, plasma [Na+]; R, tubular reabsorption of Na+; UNa+V, Na+ excretion rate. |
| The final component in the response to ECF volume expansion is the excretion of water. As Na+ excretion increases, plasma osmolality begins to fall. This decreases the secretion of ADH. ADH secretion is also decreased in response to the elevated levels of natriuretic peptides. In addition, these natriuretic peptides inhibit the action of ADH on the collecting duct. Together, these effects decrease reabsorption of water by the collecting duct and thereby increase excretion of water by the kidneys. Thus, excretion of Na+ and water occurs in concert; euvolemia is restored, and body fluid osmolality remains constant. The time course of this response (hours to days) depends on the magnitude of the ECF volume expansion. Hence, if the degree of ECF volume expansion is small, the mechanisms just described generally restore euvolemia within 24 hours. However, with large degrees of ECF volume expansion, the response can take several days.
|
| page 616 | | | page 617 |
| In brief, the renal response to ECF volume expansion involves the integrated action of all parts of the nephron: (1) the filtered load of Na+ is increased, (2)
reabsorption in the proximal tubule and loop of Henle is reduced (GFR is increased, whereas proximal reabsorption is decreased; thus, glomerulotubular balance does not occur under this condition), and (3) delivery of Na+ to the distal tubule is increased. This increased delivery, along with the inhibition of reabsorption in the distal tubule and collecting duct, results in the excretion of a larger fraction of the filtered load of Na+ and thus restores euvolemia.
|
| Control of NaCl Excretion with Volume Contraction
|
During ECF volume contraction, the high-pressure and low-pressure vascular volume sensors send signals to the kidneys that reduce the excretion of NaCl and water. The signals that act on the kidneys include
- Increased renal sympathetic nerve activity
- Increased secretion of renin, which results in elevated angiotensin II levels and thus increased secretion of aldosterone by the adrenal cortex
- Inhibition of ANP and BNP secretion by the heart and urodilatin production by the kidneys
- Stimulation of ADH secretion by the posterior pituitary
|
| Figure 34-9 Integrated response to ECF volume contraction. Numbers refer to the description of the response in the text. PNa+, plasma [Na+]; R, tubular reabsorption of Na+; UNa+V; Na+ excretion rate. |
| page 617 | | | page 618 |
The integrated response of the nephron to these signals is illustrated in Figure 34-9. The general response is as follows (the numbers correlate with those encircled in Fig. 34-9):
- The GFR decreases. Afferent and efferent arteriolar constriction occurs as a result of increased renal sympathetic nerve activity. The effect appears to be greater on the afferent than on the efferent arteriole. This causes hydrostatic pressure in the glomerular capillary to fall and thereby decreases the GFR. Because renal plasma flow decreases more than the GFR, the filtration fraction increases. The decrease in GFR reduces the filtered load of Na+.
- Na+ reabsorption by the proximal tubule and loop of Henle is increased. Several mechanisms augment reabsorption of Na+ in the proximal tubule. For example, increased sympathetic nerve activity and angiotensin II levels directly stimulate Na+ reabsorption. The decreased hydrostatic pressure within the glomerular capillaries also leads to a decrease in the hydrostatic pressure within the peritubular capillaries. In addition, the increased filtration fraction results in an increase in peritubular oncotic pressure. These alterations in capillary Starling forces facilitate the movement of fluid from the lateral intercellular space into the capillary and thereby stimulate the reabsorption of solute (e.g., NaCl) and water by the proximal tubule (see Chapter 33 for a complete description of this mechanism). The reduced filtered load and enhanced proximal tubule reabsorption decrease the delivery of Na+ to the loop of Henle. Increased sympathetic nerve activity, as well as elevated levels of angiotensin II
and aldosterone, stimulate reabsorption of Na+ by the thick ascending limb. Because sympathetic nerve activity is increased and angiotensin II and aldosterone levels are elevated during ECF volume contraction, increased Na+ reabsorption by this segment is expected. Thus, less Na+ is delivered to the distal tubule.
- Na+ reabsorption by the distal tubule and collecting duct is enhanced. The small amount of Na+ that is delivered to the distal tubule is almost completely reabsorbed because transport in this segment and the collecting duct is enhanced. This stimulation of Na+ reabsorption by the distal tubule and collecting duct is mainly induced by increased aldosterone levels. In addition, plasma levels of natriuretic peptides, which inhibit reabsorption in the collecting duct, are reduced.
|
| Finally, water reabsorption by the late portion of the distal tubule and the collecting duct is enhanced by ADH, levels of which are elevated through activation of the low- and high-pressure vascular volume sensors, as well as by the elevated levels of angiotensin II. As a result, water excretion is reduced. Because both water and Na+ are retained by the kidneys in equal proportions, euvolemia is reestablished, and body fluid osmolality remains constant. The time course of this expansion in ECF (hours to days) and the degree to which euvolemia is attained depend on the magnitude of the ECF volume contraction, as well as the dietary intake of Na+. Thus, the kidneys reduce excretion of Na+, and euvolemia can be restored more quickly if additional NaCl is ingested in the diet.
|
| In brief, the nephron's response to ECF volume contraction involves the integrated action of all its segments: (1) the filtered load of Na+ is decreased, (2) reabsorption by the proximal tubule and loop of Henle is enhanced (the GFR is decreased, whereas proximal reabsorption is increased; thus, glomerulotubular balance does not occur under this condition), and (3) delivery of Na+ to the distal tubule is reduced. This decreased delivery, together with enhanced Na+ reabsorption by the distal tubule and collecting duct, virtually eliminates Na+ from the urine.
|
- Regulation of body fluid osmolality (i.e., steady-state balance) requires that the amount of water added to the body exactly match the amount lost from the body. Water is lost from the body by several routes (e.g., during respiration, with sweating, and in feces). The kidneys are the only regulated route of water excretion. Excretion of water by the kidneys is regulated by ADH secreted from the posterior pituitary. When ADH levels are high, the kidneys excrete a small volume of hyperosmotic urine. When ADH levels are low, a large volume of hypoosmotic urine is excreted.
- Disorders of water balance alter body fluid osmolality. Because Na+ and its anions are the major determinant of ECF osmolality, disorders in water balance are manifested as changes in ECF [Na+]. Positive water balance (intake > excretion) results in a decrease in body fluid osmolality and hyponatremia. Negative water balance (intake < excretion) results in an increase in body fluid osmolality and hypernatremia.
- ECF volume is determined by the amount of Na+ in this compartment. To maintain constant ECF volume (i.e., euvolemia), Na+ excretion must match Na+ intake. The kidneys are the major route for regulating excretion of Na+ from the body. Volume sensors located primarily in the vascular system monitor volume and pressure. When ECF volume expansion occurs, neural and hormonal signals are sent to the kidneys to increase the excretion of NaCl and water and thereby restore euvolemia. When ECF volume contraction occurs, neural and hormonal signals are sent to the kidneys to decrease NaCl and water excretion and thereby restore euvolemia. The sympathetic nervous system, the renin-angiotensin-aldosterone system, and natriuretic peptides are important components of the system needed to maintain steady-state Na+ balance.
|
|
|