| 39 Hormonal Regulation of Calcium and Phosphate Metabolism
|
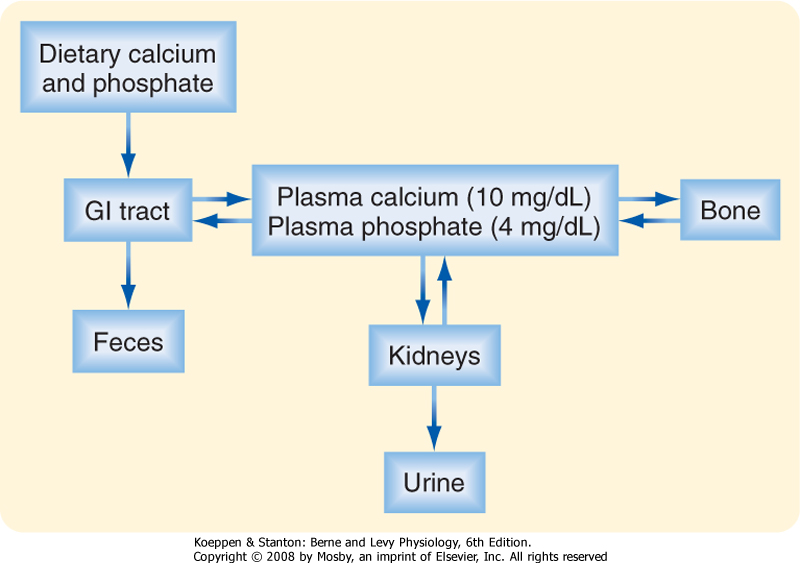
| Calcium (Ca++) and phosphate are essential to human life because they play important structural roles in hard tissues (i.e., bones and teeth) and important regulatory roles in metabolic and signaling pathways. In blood, most phosphate exists in the ionized form of phosphoric acid, which is called inorganic phosphate (Pi). The two primary sources of circulating Ca++ and Pi are the diet and the skeleton (Fig. 39-1). Two hormones, 1,25-dihydroxyvitamin D (also called calcitriol) and parathyroid hormone (PTH), regulate intestinal absorption of Ca++ and Pi and release of Ca++ and Pi into the circulation after bone resorption. The primary processes for removal of Ca++ and Pi from blood are renal excretion and bone formation (Fig. 39-1). 1,25-Dihydroxyvitamin D and PTH regulate both processes. Other hormones and paracrine growth factors also regulate Ca++ and Pi homeostasis.
|
| CRUCIAL ROLES OF CALCIUM AND PHOSPHATE IN CELLULAR PHYSIOLOGY
|
| Calcium is an essential dietary element. In addition to obtaining Ca++ from the diet, humans contain a vast store (i.e., >1 kg) of Ca++ in their bones, which can be called on to maintain normal circulating levels of Ca++ in times of dietary restriction and during the increased demands of pregnancy and nursing. Circulating Ca++ exists in three forms (Table 39-1): free ionized Ca++, protein-bound Ca++, and Ca++ complexed with anions (e.g., phosphates, HCO3-, citrate). The ionized form represents about 50% of circulating Ca++, and because this form is so critical to many cellular functions, [Ca++] in both the extracellular and intracellular compartments is tightly controlled. Circulating Ca++ is under direct hormonal control and normally maintained in a relatively narrow range. Either too little Ca++ (hypocalcemia; total serum [Ca++] below 8.5 mg/dL [4.2 mEq/L]) or too much Ca++ (hypercalcemia; total serum [Ca++] above 10.5 mg/dL [5.2 mEq/L]) in blood can lead to a broad range of pathophysiological changes, including neuromuscular dysfunction, central nervous system dysfunction, renal insufficiency, calcification of soft tissue, and skeletal pathology.
|
| Pi is also an essential dietary element, and it is stored in large quantities in bone complexed with Ca++. Most circulating Pi is in the free ionized form, but some Pi (<20%) circulates as a protein-bound form or is complexed with cations (Table 39-1). Because soft tissues contain 10-fold more Pi than Ca++, tissue damage (e.g., crush injury with massive muscle cell death) can result in hyperphosphatemia, whereupon the increased Pi complexes with Ca++ to cause acute hypocalcemia.
|
| Pi is a key intracellular component. Indeed, it is the high-energy phosphate bonds of ATP that maintain life. Phosphorylation and dephosphorylation of proteins, lipids, second messengers, and cofactors represent key regulatory steps in numerous metabolic and signaling pathways, and phosphate also serves as the backbone for nucleic acids.
|
| PHYSIOLOGICAL REGULATION OF CALCIUM AND PHOSPHATE: PARATHYROID HORMONE AND 1,25-DIHYDROXYVITAMIN D
|
| PTH and 1,25-dihydroxyvitamin D are the two physiologically most important hormones that are dedicated to maintenance of normal blood [Ca++] and [Pi] in humans. As such, they are referred to as calciotropic hormones. The structure, synthesis, and secretion of these two hormones and their receptors will be discussed first. In the following section, the detailed actions of PTH and 1,25-dihydroxyvitamin D on the three key sites of Ca++/Pi homeostasis (i.e., gut, bone, and kidney) are discussed.
|
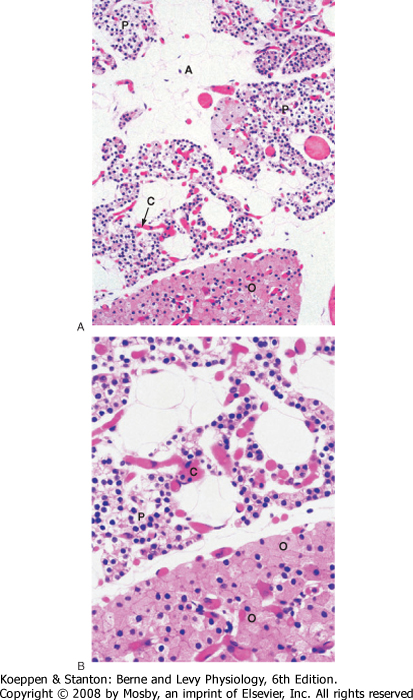
| The predominant parenchymal cell type in the parathyroid gland is the principal (also called chief) cell (Fig. 39-2).
|
| PTH is the primary hormone that protects against hypocalcemia. The primary targets of PTH are bone and the kidneys. PTH also functions in a positive feed-forward loop by stimulating production of 1,25-dihydroxyvitamin D.
|
| Structure, Synthesis, and Secretion
|
| page 696 |  | | page 697 |
| Figure 39-1 Daily Ca++ and Pi flux. |
|
Table 39-1.
Forms of Ca++ and Pi in Plasma |
| Ion | mg/dL | Ionized | Protein Bound | Complexed |
| Ca++ | 10 | 50% | 45% | 5% |
| Pi | 4 | 84% | 10% | 6% |
|
Ca++ is bound (i.e., complexed) to various anions in plasma, including HCO3-, citrate, and SO4-2. Pi is complexed to various cations, including Na+ and K+. (From Koeppen BM, Stanton BA: Renal Physiology, 4th ed. Philadelphia, Mosby, 2007.)
|
| PTH is proteolytically cleaved into biologically inactive N-terminus and C-terminus fragments that are excreted by the kidney. Older PTH assays detected both intact 1-84 PTH and inactive C-terminus fragments and therefore detected active and inactive PTH, especially in patients with renal disease. Current assays use two antibodies that recognize epitopes from both ends of the molecule, thereby more accurately measuring the intact 1-84 form of PTH. |
| PTH is secreted as an 84-amino acid polypeptide and is synthesized as a prepro-PTH, which is proteolytically processed to pro-PTH in the endoplasmic reticulum and then to PTH in the Golgi and secretory vesicles. Unlike proinsulin, all intracellular pro-PTH is normally
converted to PTH before secretion. PTH has a short half-life (<5 minutes).
|
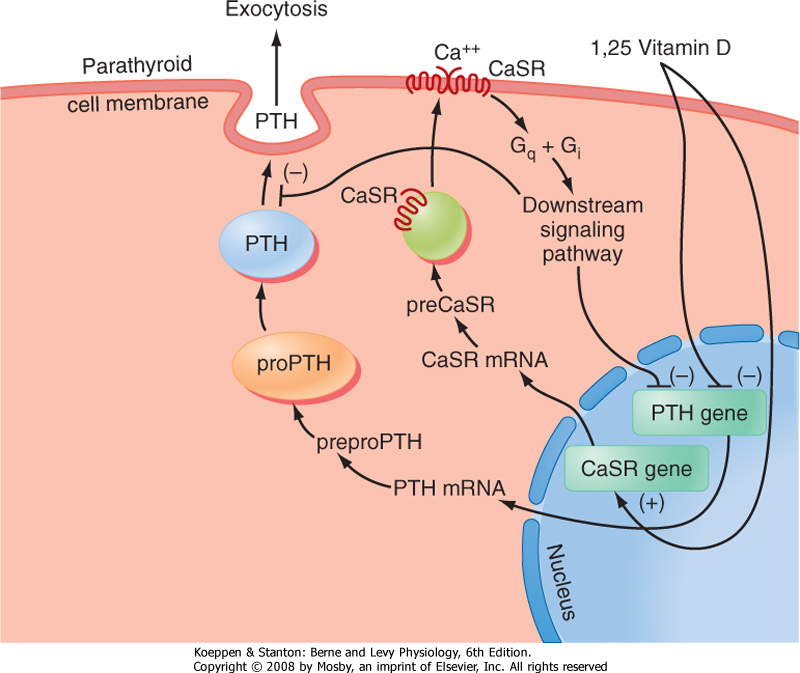
| The primary signal that stimulates PTH secretion is low circulating [Ca++] (Fig. 39-3). The extracellular [Ca++] is sensed by the parathyroid chief cell through a Ca++-sensing receptor (CaSR). In the parathyroid gland, increasing amounts of extracellular Ca++ bind to the CaSR and activate signaling pathways that repress PTH secretion.
|
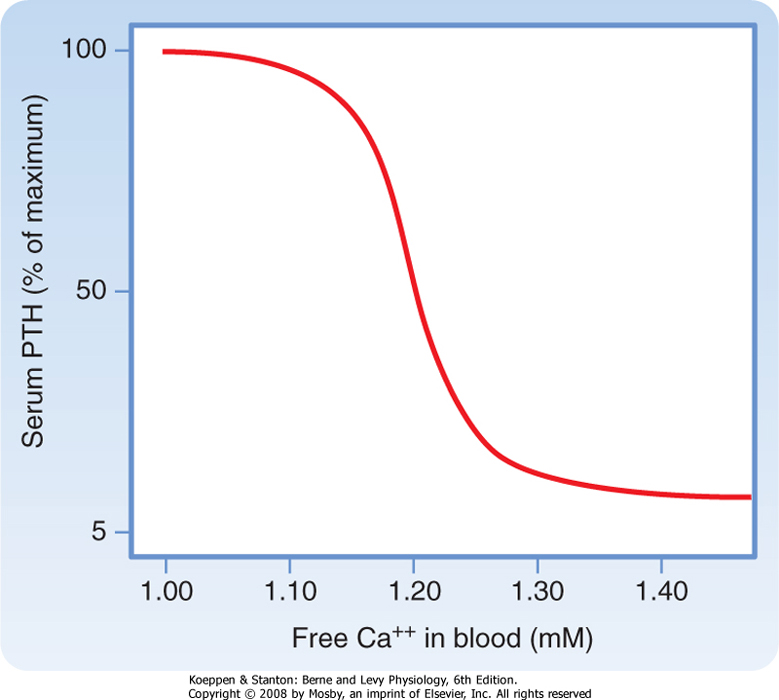
| Although the CaSR binds to extracellular Ca++ with relatively low affinity, the CaSR is extremely sensitive to changes in extracellular [Ca++]. A 0.2-mEq/L drop in blood [Ca++] produces an increase in circulating PTH levels from basal (5% of maximum) to maximum levels (Fig. 39-4). Thus, the CaSR regulates PTH output in response to subtle fluctuations in [Ca++] on a minute-to-minute basis.
|
| Figure 39-2 A and B, Histology of parathyroid glands. A, adipose tissue within parathyroid glands; C, capillaries; O, oxyphil cells; P, principal or chief cells. (From Young B et al: Wheater's Functional Histology, 5th ed. Philadelphia, Churchill Livingstone, 2006.) |
| page 697 | | | page 698 |
| Figure 39-3 Regulation of PTH gene expression and secretion. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
| Figure 39-4 Ca++/PTH secretion dose-response curve. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
| Patients with familial benign hypocalciuric hypercalcemia (FBHH) or neonatal severe hyperparathyroidism are heterozygous or homozygous, respectively, for inactivating mutations of the CaSR. In these patients the CaSR fails to appropriately inhibit PTH secretion in response to high blood levels of Ca++. The CaSR also plays a direct role in Ca++ reabsorption in the kidney. The hypocalciuria (i.e., inappropriately low Ca++ excretion in the face of high circulating [Ca++]) in patients with FBHH is due to the lowered ability of the CaSR to monitor blood calcium and respond by increasing urinary Ca++ excretion. |
|
| PTHrP is a peptide paracrine hormone produced by several tissues. PTHrP is also expressed in several developing tissues, including the growth plate of bones and in the mammary glands, and may play several roles in adults (e.g., regulation of uterine contractions). The 30 amino acids at the N-terminus of PTHrP have significant structural homology with PTH. Thus, PTHrP binds to and signals through the PTH/PTHrP receptor. PTHrP is not regulated by circulating Ca++ and normally does not play a role in Ca++/Pi homeostasis in adults. However, certain tumors secrete high levels of PTHrP, which causes hypercalcemia of malignancy and symptoms that resemble hyperparathyroidism. |
|
| PTH production is also regulated at the level of gene transcription (Fig. 39-3). The PTH gene is repressed by
a calcium response element within the promoter of this gene. Thus, the signaling pathway that is activated by binding of Ca++ to the CaSR ultimately leads to repression of PTH gene expression and synthesis. The PTH gene is also repressed by 1,25-dihydroxyvitamin D (acting through vitamin D response elements-see later). The ability of 1,25-dihydroxyvitamin D to hold PTH gene expression in check is reinforced by the coordinated up-regulation of CaSR gene expression by positive vitamin D response elements in the promoter region of the CaSR gene (Fig. 39-3).
|
| Parathyroid Hormone Receptor. Because the PTH receptor also binds PTH-related peptide (PTHrP), it is usually referred to as the PTH/PTHrP receptor. The PTH/PTHrP receptor is expressed on osteoblasts in bone and in the proximal and distal tubules of the kidney, and it is the receptor that mediates the systemic actions of PTH. However, the PTH/PTHrP receptor is also expressed in many developing organs, in which PTHrP has an important paracrine function.
|
| page 698 | | | page 699 |
| Vitamin D is actually a prohormone that must undergo two successive hydroxylation reactions to become the active form 1,25-dihydroxyvitamin D (Fig. 39-5). Vitamin D plays a critical role in Ca++ absorption and, to a lesser extent, Pi absorption by the small intestine. Vitamin D also regulates bone remodeling and renal reabsorption of Ca++ and Pi.
|
| Structure, Synthesis, and Transport of Active Vitamin D Metabolites
|
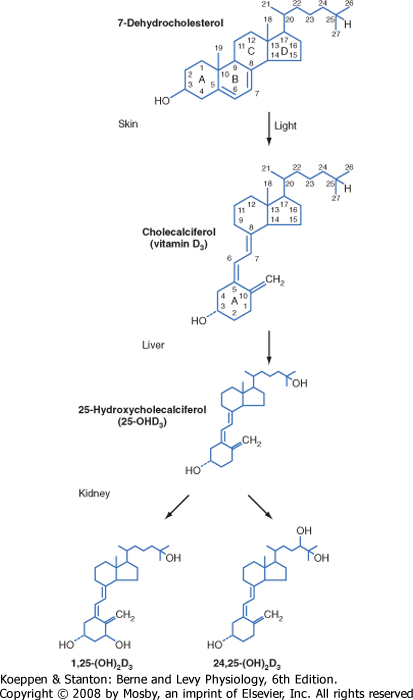
| Vitamin D3 (also called cholecalciferol) is synthesized via the conversion of 7-dehydrocholesterol by ultraviolet B light (UVB) in the more basal layers of the skin (Fig. 39-6). Vitamin D3 is therefore referred to as a secosteroid, which is a class of steroids in which one of the cholesterol rings is opened (Fig. 39-5). Vitamin D2 is produced in plants. Vitamin D3 and, to a lesser extent, vitamin D2 are absorbed from the diet and are equally effective after conversion to active hydroxylated forms. The balance between UVB-dependent, endogenously synthesized vitamin D3 and absorption of the dietary forms of vitamin D becomes important in certain situations. Individuals with higher epidermal melanin content who live in higher latitudes convert less 7-dehydrocholesterol to vitamin D3 and thus are more dependent on dietary sources of vitamin D3. Dairy products are enriched with vitamin D3, but not all individuals tolerate or enjoy dairy products. Institutionalized, sedentary elderly patients who stay indoors and avoid dairy products are particularly at risk for the development of vitamin D3 deficiency.
|
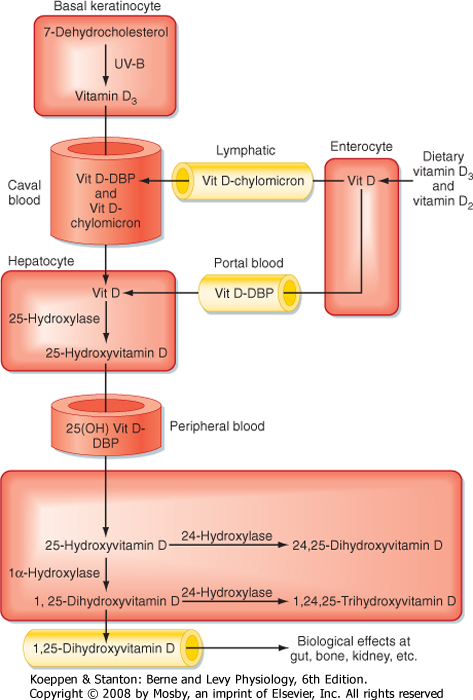
| Vitamin D3 is transported in blood from the skin to the liver. Dietary vitamin D3 and vitamin D2 reach the liver directly via transport in the portal circulation and indirectly via chylomicrons (Fig. 39-6). In the liver, vitamin D2 and vitamin D3 are hydroxylated at the 25-carbon position to yield 25-hydroxyvitamin D (at this juncture, no distinction will be made between D3 and D2 metabolites because they are equipotent). Hepatic 25-hydroxyvitamin D is expressed at a relatively constant and high level, so circulating levels largely reflect the amount of precursor available for 25-hydroxylation. Because the hydroxyl group at the 25 carbon represents the second hydroxyl group on the molecule, 25-hydroxyvitamin D is also referred to as calcifediol.
|
| 25-Hydroxyvitamin D is further hydroxylated in the proximal tubules of the kidney (Figs. 39-5 and 39-6). Hydroxylation of 25-hydroxyvitamin D in the 1 position generates 1,25-dihydroxyvitamin D, which is the most active form of vitamin D. Hydroxylation of 25-hydroxyvitamin D at the 24 position generates 24,25-dihydroxyvitamin D.
|
| Figure 39-5 Biosynthesis of 1,25-dihydroxyvitamin D. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
| Vitamin D and its metabolites circulate in blood primarily bound to vitamin D-binding protein (DBP). DBP is a serum glycoprotein that is synthesized by the liver. DBP binds more than 85% of 1,25-hydroxyvitamin D and 24,25-dihydroxyvitamin D. Because of binding to other proteins, only 0.4% of 1,25-dihydroxyvitamin D circulates as free hormone. DBP transports the highly lipophilic vitamin D in blood and provides a reservoir
of vitamin D that protects against vitamin D deficiency. DBP-bound vitamin D metabolites have a circulating half-life of several hours.
|
| page 699 | | | page 700 |
| Figure 39-6 Vitamin D metabolism. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
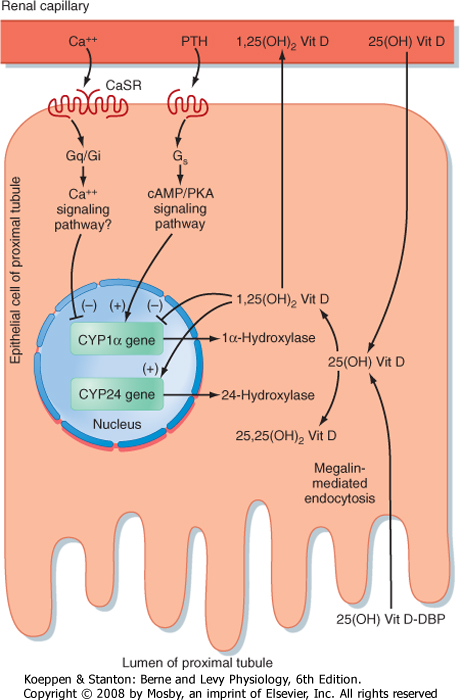
| The kidney 1α-hydroxylase enzyme (encoded by the Cyp1α gene) is tightly regulated at the transcriptional level (Fig. 39-7). 1,25-Dihydroxyvitamin D inhibits 1α-hydroxylase expression and stimulates 24-hydroxylase expression. Ca++ is also an important regulator
of renal 1α-hydroxylase. Low circulating [Ca++] indirectly stimulates renal 1α-hydroxylase expression through increased PTH levels, whereas elevated [Ca++] inhibits 1α-hydroxylase activity directly through the CaSR in the proximal tubule. A low-Pi diet also stimulates renal 1α-hydroxylase activity in a PTH-independent manner.
|
| 1,25-Dihydroxyvitamin D Receptor
|
| 1,25-Dihydroxyvitamin D exerts its actions primarily through binding to the nuclear vitamin D receptor (VDR), which is a member of the nuclear hormone receptor. The VDR is a transcription factor that binds to DNA sequences (vitamin D response elements) as a heterodimer with the retinoid X receptor (RXR). Thus, the primary action of 1,25-dihydroxyvitamin D is to regulate gene expression in its target tissues, including the small intestine, bone, kidneys, and parathyroid gland.
|
| The genomic actions of 1,25-dihydroxyvitamin D, as mediated by the VDR, occur over a period of hours or days. 1,25-Dihydroxyvitamin D also has rapid effects (seconds to 10 minutes). For example, 1,25-dihydroxyvitamin D rapidly induces absorption of Ca++ by the duodenum (transcaltachia). The VDR is also expressed in the plasma membrane of cells and is linked to rapid signaling pathways (e.g., G proteins, phosphatidylinositol-3'-kinase). Current molecular modeling has led to the development of ligands that specifically bind to the nuclear-versus the membrane-localized VDR, thus paving the way for the selective treatment of disorders related to the rapid versus slow actions of 1,25-dihydroxyvitamin D with synthetic vitamin D analogues.
|
| REGULATION OF [CA++] AND [PI] BY THE SMALL INTESTINE AND BONE
|
| An overview of the regulation of [Ca++] and [Pi] by the action of PTH and 1,25-dihydroxyvitamin D on the small intestine, bone, and parathyroid glands is summarized in Table 39-2 and in the following paragraphs. For details on the renal handling of Ca++, consult Chapter 35.
|
| page 700 | | | page 701 |
| Figure 39-7 Regulation of the renal Cyp1α gene expression by Ca++ and hormones. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
| The primary actions of calcitonin are on bone and kidney. Calcitonin lowers serum [Ca++] and [Pi], primarily by inhibiting bone resorption; however, this effect occurs only at high circulating levels. There are no complications caused by calcitonin deficiency or excess in humans. For this reason, it is unlikely that calcitonin has an important physiological role in humans. Medical interest in calcitonin stems from the fact that potent forms of calcitonin can be used therapeutically in the treatment of bone disorders. Calcitonin is also a useful histochemical marker of medullary thyroid cancer. |
| The calcitonin receptor is closely related to the PTH/PTHrP receptor. In contrast to the PTH/PTHrP receptor, the calcitonin receptor is expressed in osteoclasts. Calcitonin acts rapidly and directly on osteoclasts to suppress bone resorption. Paget's disease is characterized by excessive bone turnover that is driven by large, bizarre osteoclasts. Because these osteoclasts retain the calcitonin receptor, active forms of calcitonin can be used to suppress aberrant osteoclastic activity in patients with this disease. |
| Ca++ and Pi Transport by the Small Intestine
|
| Dietary intake of Ca++ can vary, but in general, North Americans consume about 1.5 g of Ca++ per day. Of this, about 200 mg is absorbed by the proximal part of the small intestine. Importantly, absorption of Ca++ is stimulated by 1,25-dihydroxyvitamin D, so absorption is more efficient in the face of declining dietary Ca++.
|
| page 701 | | | page 702 |
|
Table 39-2.
Actions of PTH and 1,25-Dihydroxyvitamin D on Ca++/Pi Homeostasis |
| | Small Intestine | Bone | Kidney | Parathyroid Gland |
| PTH | No direct action | Promotes osteoblastic growth and survival
Regulates M-CSF, RANKL, and OPG production by osteoblasts
Chronic high levels promote net Ca++ and Pi release from bone | Stimulates 1α-hydroxylase activity
Stimulates Ca++ reabsorption by the thick ascending limb of Henle's loop and the distal tubule
Inhibits Pi reabsorption by proximal nephrons (represses NPT2a expression) | No direct action |
| 1,25-Dihydroxyvitamin D | Increases Ca++ absorption by increasing TRPV channels, calbindin-D and PMCA expression
Marginally increases Pi absorption | Sensitizes osteoblasts to PTH
Regulates osteoid production and calcification | Minimal actions on Ca++ reabsorption
Promotes Pi reabsorption by proximal nephrons (stimulates NPT2a expression) | Directly inhibits PTH gene expression
Directly stimulates CaSR gene expression |
|
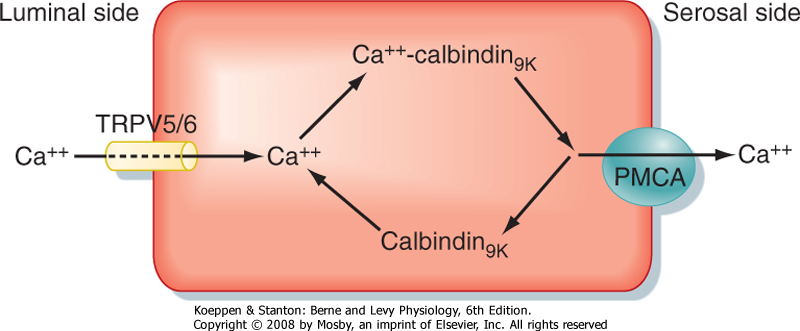
| Figure 39-8 Intestinal absorption of Ca++ via the transcellular route. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
| Ca++ is absorbed from the duodenum and jejunum by both a Ca++-regulated and a hormonally regulated transcellular route and by a passive, paracellular route. The transcellular route of Ca++ absorption is summarized in Figure 39-8. Movement of Ca++ from the lumen of the gastrointestinal tract into the enterocyte, which is favored by both chemical and electrical gradients, is facilitated by apical epithelial calcium channels called TRPV5 and TRPV6. Inside the cell Ca++ ions bind to calbindin-D9K, which maintains a low cytoplasmic [Ca++], thus preserving the favorable lumen-to-enterocyte [Ca++] gradient. Calbindin-D9K also plays a role in apical-to-basolateral shuttling of Ca++, which is transported across the basolateral membrane against an electrochemical gradient by plasma membrane
calcium ATPase (PMCA). The Na+-Ca++ sodium/calcium exchanger (NCX) also contributes to the transport of Ca++ out of enterocytes. 1,25-Dihydroxyvitamin D stimulates the expression of all the components involved in absorption of Ca++ by the small intestine.
|
| The fraction of dietary Pi absorbed by the jejunum remains relatively constant at about 70% and is under minor hormonal control by 1,25-dihydroxyvitamin D. The limiting process in transcellular Pi absorption is transport across the apical brush border, which is mediated by the Na+-Pi cotransporter (NPT2).
|
| Handling of Ca++ and Pi by Bone
|
| Bone stores a large amount of Ca++ and Pi. Once maximal bone mass has been achieved in an adult, the skeleton is constantly remodeled through the concerted activities of bone cells. The processes of bone formation (accretion) and bone resorption are in balance in a healthy, physically active, and well-nourished individual. Of the 1 kg of Ca++ immobilized in bone, about 500 mg (i.e., 0.5% of skeletal Ca++) is mobilized from and deposited in bone each day. However, the process of bone remodeling can be modulated to provide a net gain or loss of Ca++ and Pi into blood and is responsive to physical activity (or lack thereof), diet, age, and hormonal regulation. Because the integrity of bone is absolutely dependent on Ca++ and Pi, chronic dysregulation of [Ca++] and [Pi] or the hormones that regulate [Ca++] and [Pi] lead to pathological changes in bone.
|
| The process of biogenesis, growth, and remodeling of bone is complex and beyond the scope of this chapter. The key features required to understand the role of adult bone in the hormonal regulation of Ca++/Pi metabolism are discussed in this section.
|
| In adults, bone remodeling involves (1) destruction of preformed bone with the release of Ca++, Pi, and hydrolyzed fragments of the proteinaceous matrix (called osteoid) into blood and (2) new synthesis of osteoid at the site of resorption and subsequent calcification of the osteoid, primarily with Ca++ and Pi from blood. Bone remodeling occurs continually in about 2 million discrete sites involving subpopulations of bone cells called basic multicellular units.
|
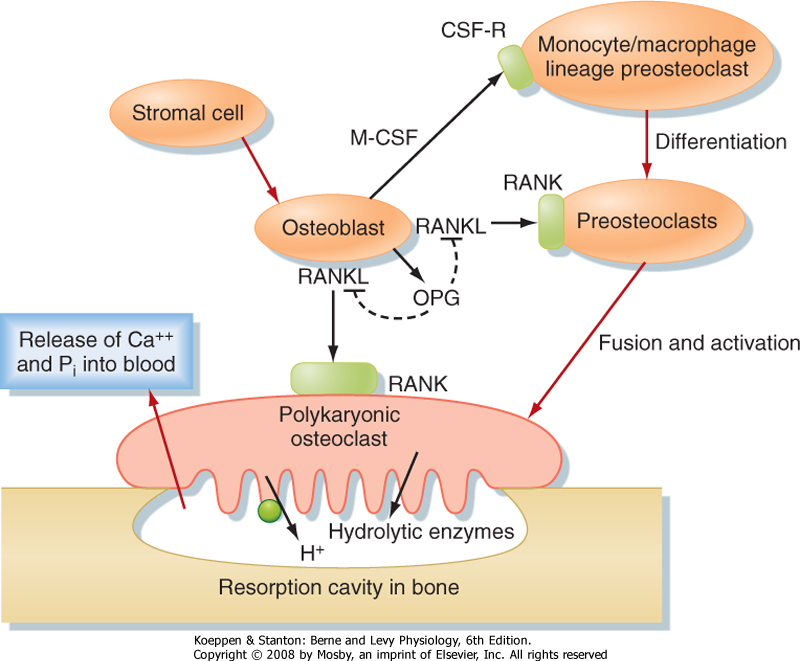
| The cells involved in bone remodeling fall into two major classes: cells that promote the formation of bone (osteoblasts) and cells that promote the resorption of bone (osteoclasts). The process of bone remodeling is a highly integrated process (Fig. 39-9). Osteoblasts express factors that induce differentiation of osteoclasts from cells of the monocyte/macrophage lineage and then fully activate osteoclast function. Osteoblasts release monocyte colony-stimulating factor (M-CSF), which induces the earliest differentiating processes that lead to osteoclast precursors. M-CSF also acts in concert with another factor, RANKL (named for receptor activator of NF-κB ligand), to promote osteoclastogenesis. RANKL binds to its receptor RANK on osteoclast precursor membranes and induces osteoclastogenesis. This process involves the clustering and fusion of several osteoclast precursors and gives rise to a fused, polykaryonic osteoclast. The perimeter of the osteoclast membrane facing the bone adheres tightly to the bone and essentially seals off the area of osteoclast-bone contact (Fig. 39-9). The osteoclast cell membrane facing the bone secretes hydrolytic enzymes and HCl. The acidic enzyme-rich microenvironment dissolves the calcified crystals, thereby releasing Ca++ and Pi into blood. After about 2 weeks, osteoclasts receive a different signal from neighboring osteoblasts. This signal is osteoprotegerin (OPG), which acts as a soluble decoy receptor for RANKL (Fig. 39-9). Consequently, the proosteoclastic signal from osteoblasts is terminated.
|
| page 702 | | | page 703 |
| Figure 39-9 Osteoblast regulation of osteoclast differentiation and function. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
| The importance of the RANK/RANKL/OPG system is underscored by mutations in the human genes for RANK and OPG, which are associated with bone deformities. Loss of RANKL in mice causes osteopetrosis (i.e., excessive bone density) because of the loss of osteoclasts. Conversely, loss of OPG causes osteoporosis (reduced bone density) because of a high number of overly active osteoclasts. |
|
| During a reversal phase, adjacent osteoblasts migrate into the resorbed area (now vacated by osteoclasts) and begin to lay down osteoid. Some of the components in osteoid promote its calcification, a process that removes Ca++ and Pi from blood. As the
osteoblasts become surrounded by and entrapped within bone, they become osteocytes that sit within small spaces, called haversian lacunae. Osteocytes remain interconnected through cell processes that run within canaliculi and form communicating junctions with adjacent cell processes. The new concentric layers of bone, along with the interconnected osteocytes and the central canal, are referred to collectively as an osteon. The exact function or functions of osteocytes are presently unclear, although evidence exists for a role of osteocytes in sensing mechanical stress in bones.
|
| In vitamin D-deficient individuals, osteoid is not properly calcified and the bone is weak. Vitamin D deficiency produces hypocalcemia and hypomagnesemia and decreases gastrointestinal absorption of Ca++ and Pi. The drop in serum [Ca++] stimulates secretion of PTH, which stimulates excretion of Pi by the kidney, thereby aggravating the loss of Pi in serum. Because the Ca++/Pi product in serum is low, bone mineralization is impaired and demineralization is increased. In children, this leads to rickets, in which the growth of long bones is abnormal and the weakened bones lead to bowing of the extremities and collapse of the rib cage. In adults, vitamin D deficiency leads to osteomalacia, which is characterized by poorly calcified osteoid associated with pain, increased risk for fracture, and vertebral collapse. The secondary elevation in PTH can produce osteoporosis. |
|
| As a calciotropic hormone, PTH is the primary endocrine regulator of bone remodeling in adults. The PTH/PTHrP receptor is expressed on osteoblasts, but not on osteoclasts. Therefore, PTH directly stimulates osteoblastic activity and stimulates osteoclastic activity indirectly through osteoblast-derived paracrine factors (i.e., M-CSF, RANKL). Intermittent administration of low doses of PTH promotes osteoblast survival and bone anabolic functions, increases bone density, and reduces the risk of fracture in humans. In contrast, sustained elevated levels of PTH shift the
balance to a relative increase in osteoclast activity, thereby increasing bone turnover and reducing bone density.
|
| Regulation of bone remodeling by PTH requires normal levels of 1,25-dihydroxyvitamin D. In vitamin D-deficient individuals, the Ca++-PTH secretion curve is shifted to the right. Thus, normal Ca++ levels are less effective in suppressing PTH secretion, and elevated PTH levels and increased bone turnover result. The VDR is expressed in osteoblasts, and normal 1,25-dihydroxyvitamin D levels are also required for coordination of osteoid production with its calcification.
|
| page 703 | | | page 704 |
| INTEGRATED PHYSIOLOGICAL REGULATION OF CA++/PI METABOLISM
|
| Response of Parathyroid Hormone and 1,25-Dihydroxyvitamin D to a Hypocalcemic Challenge
|
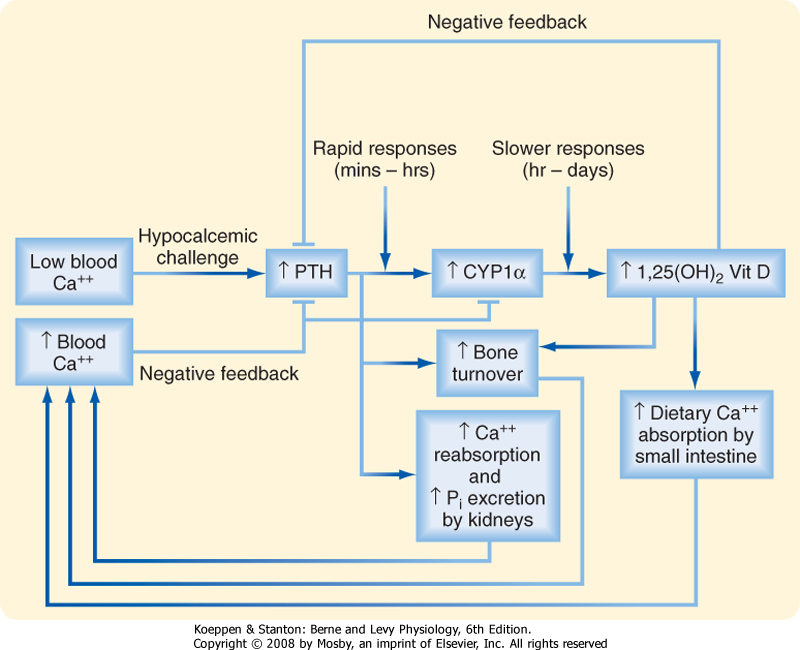
| The integrated response of PTH and 1,25-dihydroxyvitamin D to a hypocalcemic challenge is shown in Figure 39-10. Low blood [Ca++], detected by the CaSR on parathyroid chief cells, stimulates secretion of PTH. In the kidney, PTH increases Ca++ reabsorption in the thick ascending limb of Henle's loop and the distal tubule. Hypocalcemia also stimulates Ca++ reabsorption by activating the CaSR and, to a lesser extent, by increasing 1,25-dihydroxyvitamin D levels. PTH inhibits NPT2, thereby increasing excretion of Pi. The relative loss of Pi increases ionized [Ca++] in blood. In bone, PTH stimulates osteoblasts to secrete RANKL, which in turn rapidly increases osteoclast activity and leads to increased bone resorption and release of Ca++ and Pi into blood.
|
| In a slower phase of the response to hypocalcemia, PTH and low [Ca++] directly stimulate 1α-hydroxylase (CYP1α) expression in the proximal renal tubule, thereby increasing 1,25-dihydroxyvitamin D levels. In the small intestine, 1,25-dihydroxyvitamin D stimulates absorption of Ca++. These effects occur over a period of hours and days and involve increased expression of TRPV5 and TRPV6 Ca++ channels, calbindin-D9K, and PMCA. 1,25-Dihydroxyvitamin D also stimulates osteoblast release of RANKL, thereby amplifying the effect of PTH.
|
| Figure 39-10 Integrated response to a hypocalcemic challenge. (Modified from Porterfield SP, White BA: Endocrine Physiology, 3rd ed. Philadelphia, Mosby, 2007.) |
| 1,25-Dihydroxyvitamin D, along with the CaSR, plays a key role in a negative-feedback mechanism. Elevated PTH stimulates the production of 1,25-dihydroxyvitamin D,
which inhibits PTH gene expression directly and indirectly by up-regulating the CaSR. 1,25-Dihydroxyvitamin D also represses renal 1α-hydroxylase activity while increasing 24-hydroxylase activity. Thus, as blood [Ca++] returns to normal, PTH secretion and 1α-hydroxylase activity fall.
|
| Regulation by Gonadal and Adrenal Steroid Hormones
|
| Gonadal and adrenal steroid hormones have profound effects on Ca++ and Pi metabolism and on bone. Estradiol-17β (E2; see Chapter 43) has bone anabolic and calciotropic effects and stimulates intestinal Ca++ absorption. E2 is also one of the most potent regulators of osteoblast and osteoclast function. Estrogen promotes the survival of osteoblasts and apoptosis of osteoclasts, thereby favoring bone formation over resorption. In postmenopausal women, estrogen deficiency results in an initial phase of rapid bone loss that lasts about 5 years, followed by a second phase of slower bone loss that results in hypocalcemia because of inefficient Ca++ absorption and renal Ca++ wasting. This can result in secondary hyperparathyroidism, which further exacerbates the bone loss. Androgens also have bone anabolic and calciotropic effects, although some of these effects are due to the peripheral conversion of testosterone to E2 (see Chapter 43).
|
| In contrast to gonadal steroids, the glucocorticoids (e.g., cortisol) promote bone resorption and renal Ca++ wasting and inhibit intestinal Ca++ absorption. Patients treated with high levels of a glucocorticoid (e.g., as an antiinflammatory and immunosuppressive drug) can have glucocorticoid-induced osteoporosis.
|
| page 704 | | | page 705 |
| Primary hyperparathyroidism is caused by excessive production of PTH by the parathyroid glands. It is frequently caused by a single adenoma confined to one of the parathyroid glands. |
| Patients with primary hyperparathyroidism have high serum [Ca++] and, in most cases, low serum [Pi]. Hypercalcemia is a result of bone demineralization, increased gastrointestinal Ca++ absorption (mediated by 1,25-dihydroxyvitamin D), and increased renal Ca++ reabsorption. The major symptoms of the disorder are directly related to increased bone resorption, hypercalcemia, and hypercalciuria. High serum [Ca++] decreases neuromuscular excitability. People with hyperparathyroidism often show psychological disorders, particularly depression. Other neurological symptoms include fatigue, mental confusion, and at very high levels (>15 mg/dL), coma. Hypercalcemia can cause cardiac arrest and peptic ulcer formation because Ca++ increases gastrin secretion. Kidney stones (nephrolithiasis) are common because hypercalcemia eventually leads to hypercalciuria and increased Pi clearance leads to phosphaturia. The high urinary [Ca++] and [Pi] increase the tendency for precipitation of Ca++/Pi salts in soft tissues of the kidney. When serum [Ca++] exceeds about 13 mg/dL with a normal phosphate level, the Ca++/Pi solubility product is exceeded. At this level, insoluble Ca++/Pi salts form, which results in calcification of soft tissues such as blood vessels, skin, lungs, and joints. |
| People with hyperparathyroidism have evidence of increased bone turnover, such as increased urinary hydroxyproline levels, which is indicative of high bone resorptive activity. Hydroxyproline is an amino acid characteristically found in type I collagen. When the collagen is degraded, urinary excretion of hydroxyproline increases. Although hyperparathyroidism will eventually cause osteoporosis (bone loss involving both osteoid and mineral), it is not necessarily the initial symptom. However, bone demineralization is apparent. |
|
- Serum [Ca++] is determined by the rate of Ca++ absorption by the gastrointestinal tract, bone formation and resorption, and renal excretion. Serum [Ca++] is normally maintained within a narrow range.
- Serum [Pi] is determined by the rate of Pi absorption by the gastrointestinal tract, soft tissue influx and efflux, bone formation and resorption, and renal excretion. Serum [Pi] normally fluctuates over a relatively wide range.
- The major physiological hormones regulating serum [Ca++] and [Pi] are PTH and 1,25-dihydroxyvitamin D (calcitriol).
- Vitamin D is synthesized from 7-dehydrocholesterol in skin in the presence of UVB light. It is hydroxylated to 25-hydroxycholecalciferol in the liver and activated by renal 1α-hydroxylase to 1,25-dihydroxyvitamin D.
- 1,25-Dihydroxyvitamin D promotes intestinal Ca++ absorption and weakly increases Pi absorption.
- The flux of Ca++ and Pi into and out of bone is determined by the relative activities of osteoblasts versus osteoclasts.
- The PTH/PTHrP receptor is expressed on osteoblasts, not on osteoclasts. PTH promotes osteoblast differentiation, proliferation, and survival, and intermittent administration of PTH promotes bone formation.
- 1,25-Dihydroxyvitamin D binds to the VDR in osteoblasts to increase osteoblast differentiation, promote the secretion of osteoid components, and sensitize osteoblasts to PTH.
|
|
|